
August 7, 1998
MEMORANDUM
| SUBJECT: | Clarification Regarding Use of SW-846 Methods |
| FROM: | Elizabeth Cotsworth, Acting Director | s |
Matt Hale for Office of Solid Waste |
| TO: | RCRA Senior Policy Analysts Regions I - X |
It recently came to the attention of EPA's Office of Solid Waste that additional guidance is needed regarding certain methods in Update III to Test Methods for Evaluating Solid Waste, Physical/Chemical Methods (SW-846) and the use of SW-846 methods in general, in order to assure appropriate use by the laboratories and the regulated community. The purpose of this memorandum is to set forth the guidance as a clarification to SW-846 for reference and distribution to the States and to other interested parties, including laboratories and the regulated community.
SW-846 contains the analytical and test methods that EPA has evaluated and found to be among those acceptable for testing under subtitle C of the Resource Conservation and Recovery Act (RCRA). In most situations, SW-846 functions as a guidance document setting forth acceptable, although not required, methods to be implemented by the user, as appropriate, in responding to RCRA-related sampling and analysis requirements. The methods are intended to be used and modified, as needed, to promote unbiased, sensitive, precise, comparable, and specific analyses and test results. In addition, with the exception of method-defined parameters (e.g., Method 1311, the Toxicity Characteristic Leaching Procedure), SW-846 methods need not be applied in a prescriptive manner. The Agency strongly recommends that the regulated entity develop a project-specific sampling and analysis plan in conjunction with other professionals (e.g., laboratories) and the regulating authority, to address both sample collection and method application and to assure the generation of data of the appropriate quality. The Disclaimer and Chapter Two of SW-846 provide additional guidance regarding the appropriate use of SW-846 methods, and Chapter One provides guidance regarding the development of a project-specific sampling and analysis plan.
SW-846 also is a "living document" that changes over time as new information, analytical technologies, and data are developed and made available. Advances in analytical instrumentation and techniques are continually reviewed by the Agency and periodically incorporated into SW-846 to support changes in the regulatory program and to improve method performance. Update III represents such an incorporation into SW-846. The update was finalized on June 13, 1997 (62 FR 32452), and included 37 revised methods and 61 new methods. Besides providing new technologies and improved methods, the Agency strove as part of Update III to address some long-standing concerns or misconceptions regarding the use of SW-846 and its methods.
Subsequent to finalizing Update III, the Office of Solid Waste received additional public comments regarding the content of a few of the methods. The Agency reviewed the comments and determined that additional guidance regarding the subject methods would be beneficial to the regulated community and regulating authorities. The Agency notes that this guidance simply clarifies the original intent of the methods and the manual, and does not represent significant changes to the Update III methods as promulgated on June 13, 1997. In the future, the Agency plans to revise the affected SW-846 methods to include this guidance.
Attachment 1 to this memorandum contains a synopsis of the clarifications to certain portions of the following SW-846, Final Update III methods:
- Method 3550B, Ultrasonic Extraction
- Method 5021, Volatile Organic Compounds in Soils and Other Solid Matrices Using Equilibrium Headspace Analysis
- Method 5035, Closed-System Purge-and-Trap and Extraction for Volatile Organics in Soil and Waste Samples
- Method 6010B, Inductively Coupled Plasma-Atomic Emission Spectrometry
- Method 8000B, Determinative Chromatographic Separations
- Method 8082, Polychlorinated Biphenyls (PCBs) by Gas Chromatography
The methods are discussed in numerical order, as listed above. Attachment 2 to this memorandum provides a more detailed discussion of the issues surrounding the clarifications. The detailed discussions in Attachment 2 should be reviewed to fully appreciate the context on which the clarifications are based. All copies of this memorandum should be distributed with both attachments
| cc: | Michael Shapiro Barnes Johnson Key Regional RCRA Contacts RCRA Branch Chiefs Enforcement Division Directors Larry Reed, Superfund Anna Virbick, UST Walt Kovalick, TIO David Friedman, EMMC Tony Pagliaro, ACIL |
Attachment 1
Synopsis of the Clarifications to Certain Update III SW-846 Methods
July 1998
This attachment provides a synopsis of the clarifications to six methods from Update III to SW-846. The methods are discussed in numerical order and include Methods 3550B, 5021, 5035, 6010B, 8000B, and 8082. The synopsis of each method is supported by a detailed discussion in Attachment 2. The reader should review the detailed discussions of these issues, in order to more fully understand the context of these clarifications.
Synopsis of Clarifications to Method 3550B - Ultrasonic Extraction
- The Agency recommends that the statements in Sections 1.5 and 1.7 of Method 3550B regarding extraction efficiency and organophosphorus pesticides be treated as cautions, not outright prohibitions on the use of this extraction technique. The discussions of the organophosphorus pesticides issue in Method 3500B and 8141A should be treated in a similar fashion.
- The Agency recommends that analysts demonstrate the performance of any extraction technique at concentrations near those found in field samples. Such demonstrations may be performed using existing performance measures such as the initial demonstration of proficiency, laboratory control samples, and matrix spike and matrix spike duplicate pairs, as already described in SW-846.
Synopsis of Clarifications to Method 5021 - Volatile Organic Compounds in Soils and Other Solid Matrices Using Equilibrium Headspace Analysis
- The Agency emphasizes that all samplers and analysts using this procedure must exercise extreme care when collecting and analyzing samples with or without preservation. The potential for loss of target compounds is significant if samples are not handled properly.
- The Agency strongly recommends the use of the preservation options available in the method to prevent loss of target compounds.
- Sampling personnel should review the introductory text in Section 6.0 of the method and consult the appropriate laboratory personnel to ensure that the options for preservation and addition of internal standards and surrogates are carried out appropriately.
Synopsis of Clarifications to Method 5035 - Closed-System Purge-and-Trap and Extraction for Volatile Organics in Soil and Waste Samples
- The Agency recommends that all soil samples collected for volatiles be preserved in some manner, whenever possible. For low concentration samples, generally those below 200 µg/kg of volatiles, preservation is essential. For samples with higher concentrations of volatiles (e.g., greater than 200 µg/kg), the Agency recommends that unpreserved samples only be collected as a last resort, and that the rationale for not preserving the samples be clearly documented in a sampling and analysis plan that is reviewed and approved by the relevant regulatory authority.
- For samples of calcareous soils that effervesce on contact with the sodium bisulfate preservative solution, the Agency recommends that such strongly reacting samples be collected in a device such as the EnCore sampler, stored at 4°C or less, and analyzed within 48 hours of collection. Longer holding times may be implemented if it can be conclusively demonstrated that alternative preservation techniques, such as freezing samples immediately after collection (and keeping them frozen during shipping and storage), or immediately upon receipt in the laboratory, does not compromise sample integrity.
- Sample vials are weighed in the field before use. Vials containing methanol are used for high concentration samples. When the difference between the weight determined at the time that the vial was prepared in the laboratory and the weight in the field varies significantly, the vial should not be used because the difference suggests that the vial is losing methanol. If both weight measurements are made in the laboratory before use, the loss of 0.01 g or more should be used as the point at which the vial is not used. If the initial weight measurement is made in the laboratory and the subsequent weight measurement made in the field, or if both weight measurements are made in the field, the loss of 0.2 g or more should be used as the point at which the vial is not used.
- The Agency recommends that the 1:1 soil to solvent ratio be used as a
default value (e.g., 5 g of soil in a vial containing 5 mL of methanol).
Analysts wishing to employ a 1:1 soil to solvent ratio should demonstrate that
the amount of solvent is sufficient to submerge the entire plug of soil in the
vial for typical soils from the site of interest or a similar site. If the
amount of solvent is not sufficient to cover the soil plug, additional methanol
should be added to ensure that the methanol extract can be removed for purging
and that the loss of target compounds to the sample headspace will be
minimized. As already noted in Section 6.2.2.6, other sample weights and
volumes, resulting in other soil to solvent ratios, are acceptable, provided
that the overall performance of the procedure has been demonstrated to be
appropriate and appropriately documented.
Analysts and samplers should also be aware that the use of methanol preservation introduces a dilution factor that may affect the ability to determine the analytes of interest at a specific regulatory level. The dilution factor is often on the order of 50 (for a 1:1 soil to solvent ratio) or 100 (for a 1:2 soil to solvent ratio), but the exact dilution factor should be evaluated in consultation with the laboratory performing the analysis.
- With regard to the addition of surrogates to high concentration samples, analysts should follow the high concentration procedures as described in the method, ignoring the erroneous statement in the introductory text of Section 7.3 of the method.
Synopsis of Clarifications to Method 6010B - Inductively Coupled Plasma-Atomic Emission Spectrometry
- The calibration range check described in Section 7.2.5.4 employs an acceptance criterion of ±10%, allowing small excursions below and above the predicted response for the highest standard.
- As the result of an editorial error, the discussion in Section 8.6.1.2 duplicates that in Section 8.6.1.1. Only one check standard is to be run in Section 8.6.1, and evaluated using the criterion in Section 8.6.1.1.
- There is an editorial error in the text in Section 8.6.1.3. The intended
text for Section 8.6.1.3 is provided below, and will be incorporated into the
next revision of the method.
8.6.1.3 The results of the calibration blank are to be less than three times the laboratory's IDL for each analyte. If this is not the case, the reason for the out-of-control condition must be found and corrected, and the affected samples must be reanalyzed. If the laboratory consistently has concentrations greater than three times the IDL, then the IDL may be indicative of an estimated IDL and should be re-evaluated. If the blank is less than 1/10th of the concentration of the lowest sample of the batch, the analysis need not be terminated.
Synopsis of Clarifications to Method 8000B - Determinative Chromatographic Separations
- Calibrations employing least squares regressions may use a weighting factor of (1/concentration) or (1/concentration2) in place of the factor of (1/SD2) that is discussed in the method. The relative standard error (RSE) is a useful measure of the goodness of fit of a calibration model and the Agency encourages its use, employing the same numerical limits provided in many methods for the relative standard deviation (RSD). There is a typographical error in the third equation on page 21 of the method. The weighting factor (1/SD2) should not appear in that equation.
- Reporting data from dual-column chromatographic analyses is an issue that should be addressed in the sampling and analysis plan. In the absence of such a plan that discusses this issue, the Agency has provided a default approach in Method 8000B. That approach stipulates that in instances when the relative percent difference between two values exceeds 40% and there is no evidence of chromatographic anomalies or interferences, then the higher value is reported and the data user is notified of the possible problem. When interferences or anomalies are present, the analyst should take reasonable steps to resolve the problems, and the default approach provides the regulated entity with an incentive to have such problems resolved. When the difference is less that 40%, the choice of which value to report is a project-specific issue.
- Section 8.1 of Method 8000B contains an editorial error. The Agency is not
encouraging instrument-specific QC limits, but does recommend method-specific
QC limits. Each laboratory is expected to operate a formal quality assurance
program. The sentence indicating that expectation was deleted from Section 8.1
in error from the Final Update III revision. Section 8.1 should read as
follows:
8.1 Refer to Chapter One for specific quality control procedures. Each laboratory using SW-846 methods should maintain a formal quality assurance program. The development of in-house QC limits for each method is encouraged, as described in Sec. 8.7. In general, the following QC requirements pertain to all the determinative methods listed in Sec. 1.1 unless superseded by specific requirements provided in each determinative method.
- Section 8.5 of Method 8000B specifies a frequency of one MS/MSD pair for every 20 field samples. This is a default frequency that may be adjusted in the context of a sampling and analysis plan approved by the relevant regulatory authority. Further, the purpose of the MS/MSD analyses is to provide information on the applicability of the analytical method to the sample matrix. The Agency stresses that the appropriate use of MS/MSD results is to evaluate method performance in the matrix of interest, not laboratory performance.
Synopsis of Clarifications to Method 8082 - Polychlorinated Biphenyls (PCBs) by Gas Chromatography
- Section 8.3. of Method 8082 addresses the initial demonstration of proficiency. This test is to be conducted by each laboratory prior to the analysis of samples, and serves to demonstrate the laboratory's ability to perform the method in a clean matrix. Section 8.3.1.1 states that the QC reference sample be analyzed at a frequency of once for each group of up to 20 field samples. This section is clearly in error, since performance of the initial demonstration is not tied to a specific group of field samples. The specifications for the frequency of the initial demonstration are correctly provided in Section 8.3.1.
- Section 8.3.1.2 of Method 8082 provides quality control acceptance criteria of 80-120% recovery for the initial demonstration of proficiency. The Agency recognizes that these limits conflict with the more general guidance provided in Method 8000 regarding the initial demonstration. As specifically described in Method 8000, limits of 70-130% recovery should be used by the laboratory as interim guidance while the laboratory collects enough data to generate in-house control limits for the initial demonstration of proficiency. Once such limits are generated, the 70-130% guidance limits no longer apply. The Agency recommends that analysts use the approach described in Method 8000 in place of the discussion of Section 8.3.1.2 in Method 8082.
Attachment 2
Detailed Discussion of the Clarifications to Certain Update III SW-846
Methods
July 1998
This attachment contains a detailed discussion of the clarifications to certain portions of the following SW-846, Final Update III methods:
- Method 3550B, Ultrasonic Extraction
- Method 5021, Volatile Organic Compounds in Soils and Other Solid Matrices Using Equilibrium Headspace Analysis
- Method 5035, Closed-System Purge-and-Trap and Extraction for Volatile Organics in Soil and Waste Samples
- Method 6010B, Inductively Coupled Plasma-Atomic Emission Spectrometry
- Method 8000B, Determinative Chromatographic Separations
- Method 8082, Polychlorinated Biphenyls (PCBs) by Gas Chromatography
Method 3550B - Ultrasonic Extraction
Section 1.5 of Method 3550B states that ultrasonic extraction may not be appropriate for the extraction of organophosphorus pesticides (OPPs). Section 1.7 of the method states that the method is not appropriate for applications which require high extraction efficiency for all analytes at very low concentrations. The Agency recognizes that these statements and similar discussions of the OPP issue in Methods 3500B and 8141A have raised concerns among laboratories and regulators alike.
The Agency added these statements to Method 3550B in response to long-standing concerns about the extraction efficiency of the technique in general, and based on data on OPPs that were generated in 1987 in connection with Method 8140. The Agency has performed an extensive review of a variety of studies, some published in the open literature and some conducted by Agency researchers or under contract to the Agency. Based on that review, there are few well-designed studies comparing the use of ultrasonic extraction to any other extraction technique that address what the Agency considered "very low concentrations" when it added the language to Method 3550B. A few of the published studies have looked at concentrations as low as 100 µg/kg, but most of the studies have been performed on samples containing part per million (ppm or mg/kg) concentrations. In addition, even the best of these studies were not designed to permit meaningful statistical evaluations of the results. For example, several studies compared duplicate or sometimes triplicate ultrasonic extractions to single extractions by another technique. Several of the studies used different solvents when comparing different techniques, such that the effect of the solvent cannot be distinguished from the effect of the extraction technique itself. In other studies, the spiking levels were not consistent across the methods that were evaluated. The end result of these shortcomings is that, while the results for ultrasonic extraction may "look" better or worse than another technique to some observers, meaningful statistical comparisons cannot be performed.
Extraction techniques such as ultrasonic extraction employ relatively large volumes of organic solvents. For the past several years, the Agency as a whole has been working to reduce the use of solvents in its own laboratories and in the analytical methods associated with its various regulatory programs.
Therefore, in response to the concerns raised about this method, the Agency wishes to clarify its intentions with regard to ultrasonic extraction. First, the Agency recommends that the analyst demonstrate that any extraction technique is effective for the analytes of interest, at the levels of interest, in the matrix of interest. Many regulatory limits associated with the RCRA program are in the high part per billion (ppb) range or higher. However, the Agency recognizes that many analyses are performed to determine the concentrations of analytes of interest that are present in the low part per billion range and even the sub part per billion range. In those cases, the performance demonstration should focus on similar low ppb levels. The performance demonstrations involved are those already described in the SW-846 methods, and include the initial demonstration of proficiency, the laboratory control sample, and the matrix spike and matrix spike duplicate.
The Agency also acknowledges that its initial concerns about the use of ultrasonic extraction for OPPs may be due to other causes. In the next revision of the methods, the Agency plans to remove the language in question from Sections 1.5 and 1.7 of Method 3550 and corresponding language in Method 3500. Until that time, the Agency advises analysts to view the current language as a caution, not a prohibition, regarding the use of this technique and to use the existing performance measures described in SW-846 to demonstrate the performance of any extraction technique at the levels of interest for a specific project. The Agency plans to add language to Method 3500 stressing the importance of demonstrating the performance of any extraction technique at concentrations relevant to the analysis of field samples.
Method 5021 - Volatile Organic Compounds in Soils and Other Solid Matrices Using Equilibrium Headspace Analysis
Sections 6.1, 6.3, 7.4.2.3, and 7.5 of Method 5021 address the collection and analysis of samples that are not preserved in the field. The text in these sections has led to concerns and/or confusion regarding the Agency's intent with regard to sample collection and preservation. These concerns have been accentuated by the fact that the equilibrium headspace procedure is less commonly used by environmental laboratories than other techniques for the preparation of samples for the analysis of volatile constituents.
The Agency emphasizes that all samplers and analysts using this procedure must exercise extreme care when collecting and analyzing samples with or without preservation. The potential for loss of target compounds is significant if samples are not handled properly. The Agency strongly recommends the use of the preservation options available in the method to prevent loss of target compounds. EPA also plans to revise parts of Sections 6 and 7 in a future revision of the method to clarify these issues. Sampling personnel should review the introductory text in Section 6.0 of the method and consult the appropriate laboratory personnel to ensure that the options for preservation and addition of internal standards and surrogates are carried out appropriately.
Method 5035 - Closed-System Purge-and-Trap and Extraction for Volatile Organics in Soil and Waste Samples
Item 1 - Preservation of High Concentration Samples
Sections 6.1.2, 6.2.3, and 7.3.1 of Method 5035 address the collection and analysis of high concentration samples that are not preserved in the field. The text in these sections has led to concerns and/or confusion regarding the Agency's intent in Method 5035. By way of clarification, the Agency stresses that these sections describe only those samples that can reasonably be expected to contain greater than 200 µg/kg of volatile target compounds. The discussion must not be construed to suggest that low concentration soil and waste samples (e.g., those with less than 200 µg/kg) should be collected without the use of some preservative.
Under the best of circumstances, all samples, including those with high concentrations of volatile constituents, should be preserved in some manner. However, the preservation techniques are expected to differ for different concentration ranges and with different determinative methods. Collecting samples in vials containing an aqueous sodium bisulfate solution and analyzing them using the closed-system purge-and-trap described in Method 5035 represents one such preservation approach. However, this approach is not practical for samples containing concentrations of analytes over the calibration range of the determinative technique, since the sample cannot be diluted, nor can a smaller sample aliquot be removed from the original sample vial without the loss of volatile constituents.
Given that the purge-and-trap device used in Method 5035 is a closed-system, it is not possible to dilute the original sample when the observed concentration exceeds the calibration range of the method. The 200 µg/kg cutoff that distinguishes "low concentration" from "high concentration" samples is based on the commonly used upper limit of the calibration range for Method 8260, a GC/MS procedure. This upper limit is somewhat instrument-dependent. Some laboratories may be able to establish a linear range that exceeds 200 µg/kg on some instruments. The upper limit is also method-dependent, and other determinative methods that may be used in conjunction with Method 5035 may have different calibration ranges compared to Method 8260. Thus, there may be situations in which samples with less than 200 µg/kg of any volatile analyte may cause difficulties for some laboratories using some determinative methods.
Method 5035 includes procedures for collecting high concentration soil and waste samples in a vial containing methanol or other water-miscible solvent as a preservative. The Agency recognizes that other preservation techniques are available or may become available in the future. For example, individuals may use sealed sampling devices, such as the EnCore sampler described in the method, to preserve high concentration samples, when appropriate. While the Agency strongly recommends that some type of preservation technique be applied whenever possible, the Agency recognizes that there are instances where preservation is not practical for such high concentration samples. In these instances, the Agency believes that the potential loss of volatiles from such high concentration samples may be an acceptable risk, provided that the samplers and the laboratory have taken reasonable steps to minimize the loss of volatiles. To that end, the Agency recommends that the collection of unpreserved samples only be considered for those materials that can reasonably be expected to contain high levels of volatiles, and that the rationale for not preserving the samples be clearly documented in a sampling and analysis plan that is reviewed and approved by the relevant regulatory authority. Further, the Agency recommends that the laboratory be consulted prior to sample collection to determine the relevant cutoff concentration for the specific determinative method to be employed. Whatever approach is employed, the ultimate data user should be advised of the preservation approach, and must be advised of those instances in which no preservation was employed.
Item 2 - Holding Time for Samples Collected in the EnCore Sampler
Section 6.2.1.8 of Method 5035 provides a 48-hour holding time for samples collected in the EnCore sampler. At the time that Method 5035 was promulgated, the Agency had insufficient data on samples collected with this device to justify a longer holding time. Since the promulgation of the method, the Agency has become aware of additional data that may indicate that a longer holding time is appropriate. For example, test data submitted to the Agency indicate that a 7-day holding time may be acceptable for six common volatile compounds using the stainless steel EnCore sampler, although the stainless steel version of the EnCore sampler is no longer manufactured. The Agency is reviewing additional holding time data for the nylon version of the EnCore sampler. If the results from this review, and the review of any subsequently developed data, are sufficient to support a longer holding time, the Agency will incorporate that change into a future method revision.
The Agency is aware that for calcareous soils, such as those found in parts of the southern U.S., the use of the sodium bisulfate preservative solution is not appropriate, since the calcium carbonate in the soil will effervesce in the presence of the preservative solution, potentially splattering the sample onto the vial threads, driving the volatile analytes out of the solution, etc. This problem is addressed in the note in Section 6.2.1.2, which recommends that samples that vigorously react with the preservative solution be collected in vials without the preservative.
This recommendation has led to concerns about the integrity of such samples. Therefore, the Agency additionally recommends that such strongly reacting samples be collected in a device such as the EnCore sampler, stored at 4°C or less, and analyzed within 48 hours of collection. Longer holding times may be implemented if it can be conclusively demonstrated that alternative preservation techniques, such as freezing samples immediately after collection (and keeping them frozen during shipping and storage), or immediately upon receipt in the laboratory, does not compromise sample integrity.
Another possible alternative to the EnCore sampler for calcareous soils is the use of glass 40 mL VOA vials containing only organic free reagent water (no acid). Samples would be collected using a cut-off syringe and placed into the VOA vials containing the water. The vials would then be frozen to preserve the samples. To prevent the glass vials from breaking due to the expansion of the water during the freezing process, the vials would be stored at an angle to allow the water to expand. The Agency has no holding time data or additional sample storage information at this time to support the use of this technique. However, this approach may be acceptable for holding times longer than 48 hours if it can be conclusively demonstrated that the technique does not compromise the integrity of the samples.
Item 3 - Weighing Sample Vials in the Field
Section 6.1.3.4 of Method 5035 contains a note that describes weighing the vials a second time after they are prepared and before they are used. The results of the second weighing are compared to the weight that was determined in the laboratory in Section 6.1.1.6. As described in that note, a difference of > 0.01 g between these two weights indicates the loss of methanol from the vial (either as liquid or vapors) and indicates that such vials should not be used to collect samples.
The specification of 0.01 g was a typographical error, and should reflect a value of 0.1 g. The method developer provided the 0.1 g specification, based on performing the second weighing in the laboratory before proceeding to the field. The Agency recognizes that there is some confusion over this second weighing step and where it is to be performed. If both weight measurements are made in the laboratory before use, the loss of 0.01 g or more should be used as the point at which the vial is not used. If the initial weight measurement is made in the laboratory and the subsequent weight measurement made in the field, or if both weight measurements are made in the field, the loss of 0.2 g or more should be used as the point at which the vial is not used.
Item 4 - Dilution Factors Attendant in Methanol Preservation of Soil Samples
Section 2.2.2 of Method 5035 describes that soil and a water-miscible solvent such as methanol are combined in a 1:1 ratio (i.e., 5 g of soil in 5 mL of methanol) in the preservation of high concentration soil samples. In contrast, Section 6.1.3 of the method describes a 1:2 ratio (i.e., 5 g of soil in 10 mL of methanol). The conflicting information is compounded by a typographical error in Section 6.2.2, which states that the dilution factor resulting from the use of methanol as a preservative exceeds 1000. The correct value for the dilution factor is 100, and stems from the use of the 1:2 soil to methanol ratio from Section 6.1.3 and the addition of at most 100 µL of the methanol extract to 5 mL of reagent water for purging.
In addition, new data suggests that a 1:1 soil to solvent ratio may be appropriate when attempting to get better method sensitivity. These data were not available at the time that Method 5035 was promulgated. The Agency will contact the method developer to obtain these data and review them. The method will be revised in the future, if appropriate. The 1:1 soil to solvent ratio appears to work well for solid samples (e.g., sandy soil) that do not expand to soak up the methanol when it is added. On the other hand, many samples, such as those with a high organic content, may expand and soak up all the free methanol, making it impossible to remove methanol extract from the sample container for purging purposes. Moreover, if the solvent does not cover all of the soil, volatile analytes will escape into the headspace and not be captured in the aliquot of solvent removed from the vial for analysis.
While the Agency regrets the conflicting soil to solvent ratios in Section 2.2.2 and 6.1.3, the method does indicate in Section 6.2.2.6 that other sample weights and volumes of methanol may be employed, "provided that the analyst can demonstrate that the sensitivity of the overall analytical procedure is appropriate for the intended application." This should include ensuring that:
- the volume of solvent used is sufficient to cover the sample solids, and
- the analyst accurately calculates and accounts for the actual soil to solvent ratio used in all subsequent calculations.
Thus, the Agency recommends that the 1:1 soil to solvent ratio be used as a default value. Analysts wishing to employ a 1:1 soil to solvent ratio should demonstrate that the amount of solvent is sufficient to submerge the entire plug of soil in the vial for typical soils from the site of interest or a similar site. If the amount of solvent is not sufficient to cover the soil plug, additional methanol should be added to ensure that the methanol extract can be removed for purging and that the loss of target compounds to the sample headspace will be minimized. The Agency notes that such a demonstration need not be made for every site or soil matrix, but that the soil to solvent ratio be considered at some point prior to the collection of samples.
The Agency also wishes to point out that the dilution factor discussion in Section 6.2.2 should be considered in the context of the combination of sampling, preparative, and determinative procedures employed for each project. Analysts and samplers should be aware that the use of methanol preservation introduces a dilution factor that may affect the ability to determine the analytes of interest at a specific regulatory level. That dilution factor is often on the order of 50 (for a 1:1 soil to solvent ratio) or 100 (for a 1:2 soil to solvent ratio), but the exact dilution factor should be evaluated in consultation with the laboratory performing the analysis.
Item 5 - Discrepancy Regarding the Addition of Surrogates
In reviewing Method 5035, the Agency noted a discrepancy in the introductory text of Section 7.3. That text summarizes the high concentration sample preparation procedure and states that an aliquot of the sample extract is added to reagent water containing the surrogates and, if applicable, the internal standards. That introductory text is in error, since the detailed description of the procedure provided in the subsequent sections, particularly 7.3.3. and 7.3.4, clearly states that the surrogates are added to the vial containing the soil sample. The Agency regrets this error and recommends that the analyst follow the specific procedures described in the method and ignore the discrepancy in the introductory text.
Method 6010B - Inductively Coupled Plasma-Atomic Emission Spectrometry
Item 1 - Calibration Range Check Procedures
Section 7.2.5.4 of Method 6010B describes the procedures to be employed to establish the upper limit of the dynamic range of an ICP instrument employed for metals analyses. As described in that section, a minimum of three, and preferably five, standards at different concentrations are analyzed. One of those standards should be near the upper limit of the dynamic range. The Agency recognizes that there is some confusion regarding the application of the QC acceptance criterion of 10% that is provided in Section 7.2.5.4 and wishes to clarify its intentions.
The Agency's original concerns stem from the often observed behavior of ICP instruments wherein the instrument response begins to level off at higher concentrations of the analytes of interest. Therefore, Section 7.2.5.4 instructs the analyst to run the series of three to five standards to establish the linear range. The analyst is expected to use the responses from all but the highest standard to construct a linear calibration line. The line then is extrapolated past the concentration of the highest standard. The results from the highest standard are compared to the extrapolated line, in order to determine if the calibration is sufficiently linear up to the concentration of that highest standard. The Agency established an acceptance criterion of 10% for the difference between the predicted (extrapolated) and observed responses for that highest standard. The Agency intended the criterion to be ±10% of the predicted response, thereby allowing small excursions below and above the prediction. The Agency intends to clarify its intentions in a subsequent revision of the method and recommends that analysts employ the ±10% criterion.
Item 2 - Use of Calibration Verification Check Standards
Section 8.6.1 of Method 6010B describes the use of several types of standards to check or verify both the initial calibration of an instrument and the continuing (daily) calibration. Section 8.6.1.2 discusses the comparison of a "check standard" to its expected value. The Agency is aware that there are concerns about the meaning of this section, particularly in relation to the text in Section 8.6.1.1. At issue is the suggestion that Section 8.6.1.2 requires that an additional check standard be analyzed beyond that which is already discussed in Section 8.6.1.1.
This is not the Agency's intention. The Agency realizes that this confusion is the result of an editorial error in the Update III version of the method, in that the two sections describe the same check of the calibration. As a result, the Agency recommends that the analysts run the check standard described in Section 8.6.1 itself, following either an initial or a daily calibration. The acceptance criterion provided in Section 8.6.1.1 should be used to evaluate the check standard results and the text in Section 8.6.1.2 can be logically ignored. The Agency plans to correct this editorial error in the next revision of the method.
Item 3 - Evaluation of the Calibration Blank
Section 8.6.1.3 of Method 6010B describes the evaluation of the calibration blank that is analyzed immediately after the daily calibration, after every 10 samples, and at the end of the analytical run, as specified in Section 8.6.1. In response to public comments received in conjunction with the proposal of Update III, the Agency indicated its intent to revise the specific text in Section 8.6.1.3. Unfortunately, the changes were not included in the final Update III version of the method.
The intended text for Section 8.6.1.3 is provided below, and will be incorporated into the next revision of the method.
8.6.1.3 The results of the calibration blank are to be less than three times the laboratory's IDL for each analyte. If this is not the case, the reason for the out-of-control condition must be found and corrected, and the affected samples must be reanalyzed. If the laboratory consistently has concentrations greater than three times the IDL, then the IDL may be indicative of an estimated IDL and should be re-evaluated. If the blank is less than 1/10th of the concentration of the lowest sample of the batch, the analysis need not be terminated.
Method 8000B - Determinative Chromatographic Separations
Item 1 - Other Measures of Goodness of Fit for Non-Linear Calibrations
A major aspect of the revisions to Method 8000B for Update III was to address the use of calibration models other than the traditional "linear, through the origin" approach that has historically been specified in EPA methods. Based on comments received during the proposal of Update III, the Agency included a wide array of possible calibration options and used several measures of the "goodness of fit" of the calibration relationship to the actual calibration data. At least one of those approaches employed the use of multiple analyses of each calibration standard, in order to develop a statistically-based calibration model. Specifically, that model used the inverse of the variance (or the standard deviation squared) as a weighting factor for a weighted least squares regression.
Based on comments received subsequent to the promulgation of Update III, EPA has reviewed the use of other specific weighting factors for a least squares regression model. Using statistical methods, it can be shown that the variance of replicate measurements within the calibration range of an instrument is approximately proportional to the concentration at each point at which the variance can be measured. As a result, it is statistically possible to use either the inverse of the concentration or the inverse of the concentration squared as a weighting factor. This approach has the advantage of not requiring multiple analyses of each calibration standard and is also supported by many laboratory data systems.
Therefore, the Agency recommends that when a weighted least squares regression is performed on the results of single injections of a multiple point calibration (e.g., not replicate injections) that the term 1/concentration or 1/(concentration)2 be used as a weighting factor. The Agency further recommends the use of this form of weighted regression over the use of an unweighted regression.
Further, the Agency recognizes that the relative standard error (RSE) is a useful measure of the goodness of fit of a calibration model that the Agency had not previously considered. The RSE is useful for both linear regression models as well as non-linear models, as it considers the error at each point in the calibration model as a function of the concentration of that standard. The RSE is calculated as shown below:
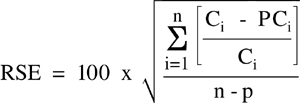
where:
n=Number of calibration points
p=Number of parameters in the model (1 for linear through the origin, 2 for
linear not through the origin, 3 for quadratic, etc.)
Ci =True concentration of the standard at level i
PCi=Predicted concentration at level i, using the calibration model
chosen
Using the RSE as a metric has the added advantage of allowing the same numerical standard to be applied to the calibration model, regardless of the form of the model. Thus, if a method states that the RSD should be < 20% for the traditional linear model through the origin, then the RSE acceptance limit can remain 20% as well. Similarly, if a method provides an RSD acceptance limit of 15%, then that same figure can be used as the acceptance limit for the RSE.
The Agency also recognizes that there is a typographical error in one of the equations in Section 7.5.2, which addresses the use of the weighted least squares regression. The weighting factor should not appear in that equation. The text and the third equation on page 21 should read as follows:
When a weighted linear least squares regression is employed, the regression equation becomes:
y=ax + b
Item 2 - Reporting of Results from Dual-Column Analyses
Section 7.10.4 of Method 8000B addresses the reporting of results when analyses are performed on two chromatographic columns of dissimilar phase. The Agency's intent with regard to this issue was to provide guidance to the analyst in those instances when an analysis necessarily produces two numerical results. The Agency is aware that this section has led to substantial confusion and wishes to point out that, as written, the discussion only applies to those instances in which the results of the two analyses differ by more than 40% and when no interferences or chromatographic anomalies are evident. The Agency's intent was to prompt the analyst to identify those instances in which the difference was relatively large (e.g., >40% RPD) and then determine if corrective action was necessary.
While the Agency agrees that, in many instances, there may be positive interferences in gas chromatographic analysis in particular that will lead to such large differences, the Agency believes that in those instances, if the interference is apparent to the analyst, then, by definition, the reporting guidance in Section 7.10.4 does not apply. However, in other instances where the interference is not apparent, the Agency believes that it is incumbent upon the analyst and the regulated entity to employ an analytical method (or methods) that is capable of determining the analyte in question without differences of this magnitude. The Agency does not believe that it is reasonable to always accept the lower of two values as the "correct" one. The Agency is not suggesting that the analyst search for a method that produces a result predetermined by the regulated entity. Rather, the Agency recognizes that some analytes are more difficult to determine in some matrices and that remedying the situation may require changes in the extraction procedures or conditions, the use of specific cleanup techniques, or the use of another determinative method.
The Agency recognizes that there are instances in which an approach other than that described in Section 7.10.4 may be appropriate. There are also reporting considerations that may apply even when the numerical differences are not so large. Therefore, the Agency recommends that data reporting in general, and reporting of dual-column results in particular, be specifically addressed in a sampling and analysis plan that is reviewed and approved by the relevant regulatory authority. However, in the absence of such a plan, the Agency believes, as stated in Section 7.10.4, that an approach that is conservative relative to environmental protection is to report the higher of the two values when the relative percent difference is greater than 40% and no interferences or chromatographic anomalies are evident.
The Agency also notes that Section 8.2.3 of Method 8000B provides a short list of potential chromatographic problems to be considered in evaluating sample results, including those with large numerical differences. In addition to those listed, the Agency recognizes that other problems will certainly occur. Examples include: one peak barely discernible above the instrumental noise and a large Gaussian peak on the other column that is most likely just an interference.
Item 3 - Instrument-Specific QC Limits
In responses to comments on the proposal of Method 8000B in Update III, the Agency agreed with the commenters that the reference to "instrument-specific" QC limits in Section 8.1 of the method was not necessary and the Agency agreed to remove the sentence that encouraged the development of such limits. An editorial error occurred in the final version of Method 8000B that retained the instrument-specific QC limit language and unintentionally deleted other important language. Therefore, the Agency is clarifying its original intent, developed in response to the Update III comments. Section 8.1 of Method 8000B should read as follows:
8.1 Refer to Chapter One for specific quality control procedures. Each laboratory using SW-846 methods should maintain a formal quality assurance program. The development of in-house QC limits for each method is encouraged, as described in Sec. 8.7. In general, the following QC requirements pertain to all the determinative methods listed in Sec. 1.1 unless superseded by specific requirements provided in each determinative method.
Item 4 - The Appropriate Use of Matrix Spike Results
Section 8.5 of Method 8000B recommends that a matrix spike (MS) and matrix spike duplicate (MSD) pair be analyzed with each batch of up to 20 samples. The MS/MSD results are an important measure of the performance of the method relative to the specific sample matrix of interest. The Agency believes that such a demonstration is an important aspect of an overall quality assurance program, and is particularly important for the RCRA program, where a wide range of different matrices are subject to regulation.
The 1 per 20 (5%) frequency is a default value that has been used in many EPA programs for many years. The Agency believes that a default frequency is needed to preclude some laboratories from deciding that no MS/MSD results need to be provided at all. However, the Agency also recognizes that other frequencies may be appropriate under other circumstances. For example, in the case of a long-term monitoring project involving a small number of analyses of a sample matrix that does not change, it should not be necessary to prove that the method applies to the matrix each time that samples are collected and analyzed.
To that end, the Agency recommends that, if another frequency for the MS/MSD analyses is chosen, that it be clearly documented in a sampling and analysis plan that is reviewed and approved by the relevant regulatory authority.
The Agency also is aware that some clients do not provide laboratories with additional volume of sample from which to prepare the MS/MSD aliquots. In some cases, the problem is an oversight on the part of the samplers. It may also be due to difficulties in obtaining sufficient volume, such as from a poorly producing groundwater well. However, in other instances, the client simply may be assuming that the laboratory will prepare the MS/MSD from another sample prepared at the same time. Unfortunately, this latter situation can result in the provision of MS/MSD results from a matrix that is only marginally related to the samples in question.
Due to the importance of the relationship between the matrices of the MS/MSD and the field samples, the Agency stresses that an MS/MSD pair (or a spiked sample and a duplicate sample) should be prepared from additional volumes of the material collected from the site in question. Each MS/MSD will require that additional sample volume from the site be provided to the laboratory by the field sampling personnel. The Agency further recommends that data users should be routinely provided with the MS/MSD results from only those QC samples associated with the field samples from the same site.
Finally, the Agency is aware of some persistent misunderstandings about the intended role of the MS/MSD analyses. The Agency stresses that the primary purpose of these QC analyses is to establish the applicability of the overall analytical approach (e.g., preparative, cleanup, and determinative methods) to the specific sample matrix from the site of interest. Unfortunately, some may believe that the MS/MSD results can and should routinely be used to evaluate performance of an individual laboratory. The Agency stresses that such use is not the Agency’s intent in specifying that MS/MSD analyses be performed at a 5% frequency. The Agency specifically included a discussion of the use of a laboratory control sample (LCS) in Method 8000B, as one tool that should be used to evaluate laboratory performance. Section 8.5.5 of Method 8000B addresses the use of LCS results in conjunction with MS/MSD results to separate issues of laboratory performance and "matrix effects."
The Agency does believe that consistent trends in MS/MSD results can be somewhat useful in evaluating laboratory performance, as are trends in surrogate recoveries, LCS recoveries, and other QC data. However, the appropriate use of a single set of MS/MSD results is to evaluate method performance in the matrix of interest, not laboratory performance.
Method 8082 - Polychlorinated Biphenyls (PCBs) by Gas Chromatography
Item 1 - Frequency of the Initial Demonstration of Proficiency
Section 8.3. of Method 8082 addresses the initial demonstration of proficiency. This test is to be conducted by each laboratory prior to the analysis of samples, and serves to demonstrate the laboratory's ability to perform the method is a clean matrix. Section 8.3.1.1 states that the QC reference sample be analyzed at a frequency of once for each group of up to 20 field samples. This section is clearly in error, since performance of the initial demonstration is not tied to a specific group of field samples. The specifications for the frequency of the initial demonstration are correctly provided in Section 8.3.1. The Agency regrets this editorial error and requests that analysts ignore this otherwise illogical discussion of frequency, which has been corrected in the Draft Update IVA revision of the method.
The Agency further notes that the discussions of calibration verification in Section 8.3.2 and 8.3.3 of Method 8082 should not be in Section 8.3, but rather should appear as part of Section 8.2, since these considerations apply to the quality control procedures necessary to evaluate GC performance that are mentioned in Section 8.2. The Agency has corrected this editorial error in the next revision of the method and advises laboratories to consider these aspects of quality control in conjunction with all GC analyses, not just the initial demonstration of proficiency described in Section 8.3.
Item 2 - QC Limits that Conflict with Method 8000
Section 8.3.1.2 of Method 8082 provides quality control acceptance criteria of 80-120% recovery for the initial demonstration of proficiency. In reviewing the method, the Agency recognizes that these limits conflict with the more general guidance provided in Method 8000 regarding the initial demonstration. As specifically described in Method 8000, limits of 70- 130% recovery should be used by the laboratory as interim guidance while the laboratory collects enough data to generate in-house control limits for the initial demonstration of proficiency. Once such limits have been generated, the 70-130% guidance limits are no longer applied.
The Agency recommends that analysts use the approach described in Method 8000 in place of the discussion of Section 8.3.1.2 in Method 8082, and has addressed this issue in the next revision of the method.
http://clu-in.org/products/regs/sw846mem.htm