Gas Chromatography
- Direct-Push Technologies
- Explosives
- Fiber Optic Chemical Sensors
- Gas Chromatography
- Geophysical Methods
- High-Resolution Site Characterization (HRSC)
- Immunoassay
- Infrared Spectroscopy
- Laser-Induced Fluorescence
- Mass Flux
- Mass Spectrometry
- Open Path Technologies
- Passive (no purge) Samplers for Groundwater
- Test Kits
- X-Ray Fluorescence
Introduction
Chromatography is the science of separation which uses a diverse group of methods to separate closely related components of complex mixtures. During gas chromatographic separation, the sample is transported via an inert gas called the mobile phase. The mobile phase carries the sample through a coiled tubular column where analytes interact with a material called the stationary phase. For separation to occur, the stationary phase must have an affinity for the analytes in the sample mixture. The mobile phase, in contrast with the stationary phase, is inert and does not interact chemically with the analytes. The only function of the mobile phase is to sweep the analyte mixture through the length of the column. Gas chromatography (GC) can be divided into two categories: (1) gas-solid and (2) gas-liquid chromatography. Gas-liquid GC, developed in 1941, is the primary GC technique used for environmental applications. Gas-solid GC is not widely used in the environmental field.
The stationary phase is chosen so that the components of the sample distribute themselves between the mobile and stationary phase to varying degrees. Those components that are strongly retained by the stationary phase move slowly relative to the flow of the mobile phase. In contrast, components that have a lower affinity for the stationary phase travel through the column at a faster rate. As a consequence of the differences in mobility, sample components separate into discrete bands that can be analyzed qualitatively and quantitatively.
Gas chromatography is the most widely used chromatographic technique for environmental analyses, and is used onsite in field investigations and by offsite reference laboratories. Field GC can provide "real-time", or near real-time data, facilitating decision making and reducing the length of field mobilization.
Typical Uses
GC analysis is a widely used technique for the field-based analysis of organic compounds present in a variety of matrices such as water, soil, soil gas, and ambient air. Typical uses include:
- Site characterization
- Stationary source testing and monitoring
- Determining the personal protective equipment (PPE) level required at hazardous waste sites
- Fence line monitoring during removal or remediation activities
- Emergency response testing
GC analysis with photoionization detection (PID) has been used extensively to characterize and remediate sites contaminated with volatile organic chemicals (VOCs). Likewise, gas chromatographs coupled with an electron capture detector are used for analysis on sites contaminated with halogenated compounds such as polychlorinated biphenyls (PCBs) or chlorinated pesticides . The recent development of truly field portable quadrupole mass spectrometers now permits GC-mass spectrometry (MS) analysis which provides definitive identification of organic compounds. The use of GC/MS is described in the Mass Spectrometry section.
Some U.S. Environmental Protection Agency (EPA) SW-846 methods can be modified for use with field GC. SW-846 methods that have been used in the field include:
- Method 8015C Non-halogenated Organics by Gas Chromatography
- Method 8015C Non-halogenated Organics by Gas Chromatography Using GC/FID
- Method 8021B Aromatic and Halogenated Volatiles by Gas Chromatography Using Photoionization and/or Electrolytic Conductivity Detectors
- Method 8081B Organochlorine Pesticides by Gas Chromatography
- Method 8082A Polychlorinated Biphenyls by Gas Chromatography
- Method 8100 Polynuclear Aromatic Hydrocarbons
Although these instrumental methods have been used in the field as in the lab, in most instances the extraction methods used to prepare the sample for analysis are different from typical lab extraction methods. Note that SW-846 extraction methods are separate methods that are not included in the instrumental methods. Instrument methods are referred to as "determinative methods" in SW-846, and the organic GC methods are in the 8000 series. SW-846 extraction methods for organics are found in the 3500 series. Methods that are less time and equipment intensive are favored for field analysis. Many of the standard extraction methods described in SW-846 for semivolatile organic compounds (SVOCs) such as polycyclic aromatic hydrocarbons (PAHs), PCBs and pesticides are best suited to fixed laboratory use as they require the use of a fume hood, large quantities of solvents, water baths and glassware that needs a time-consuming cleaning process before the glassware can be reused. The Spittler soil extraction for PCBs and other semivolatile compounds is suitable for field use, and notes on this method may be found in US EPA Field Screening Methods Catalog User's Guide EPA/540/2-88/005 September 1988.
However, the use of a thermal desorption unit built into, or as an "add on" accessory for the GC system can eliminate the need for a solvent-based, extensive sample preparation for solid samples intended for SVOC analysis. Solid-phase microextraction (SPME) can be employed in the field for VOC and SVOC analysis of water and air samples, as some field transportable GC instruments can be equipped with SPME desorption capability. SPME does not require the use of solvents. Flash vaporization has been used to introduce environmental samples contaminated with nitroaromatic explosives into a GC system, after a simple acetone extraction.
Theory of Operation
The theory of separation by GC is relatively simple and understanding the factors that affect separation allows more effective applications of GC analysis in the field. The purpose of separation is to allow identification and quantitation of individual components of a mixture and the theory of separation is detailed below. In addition to separation, detection of analytes after separation, which is an essential but separate aspect of chromatography, is presented in the section describing system components. The basic components of a simple gas chromatographic system include: (1) a carrier gas supply, (2) a syringe for sample introduction, (3) the injection port, (4) the column and oven, (5) the detector and data collection system. Components of a gas chromatograph are presented in greater detail in the section describing the system components. Schematic diagrams and photographs of instruments can also be accessed through the Systems Components section.
Before separation occurs in the chromatographic column, the mixture of components in the sample is introduced into the chromatograph through the injection port with a syringe. At this point, the analytes are vaporized (if not already in the gas phase) by the high temperature maintained in the injection port. The analytes are kept in the gaseous state by maintaining all elements of the instrument at a temperature above the boiling point of the analytes. The gas phase analytes are then immediately swept onto the chromatographic column by the mobile phase. The mobile phase is comprised of an inert carrier gas, which usually is nitrogen, helium, or hydrogen.
As the analytes are swept through the column by the mobile phase, separation occurs based on the affinity of each analyte for the stationary phase. The gas chromatographic column is composed of a coiled, tubular column and the stationary phase within the tube. GC columns are either packed or open-tubular (capillary column). Early GC columns were packed with carbon or diatomaceous earth based solids which acted as the stationary phase. In modern open-tubular columns, the stationary phase is a liquid organic compound that is coated on the internal surface of the fused silica column. Polarities of the analytes dictate the choice of stationary phase. Components of the mixture with a high degree of affinity for the stationary phase are strongly retained while components with low affinity for the stationary phase migrate rapidly through the column. As a consequence of the differences in mobility due to affinities for the stationary phase, sample components separate into discrete bands that can be qualitatively and quantitatively analyzed.
As individual components of the mixture elute the chromatographic column, they are swept by the carrier gas to a detector. The detector generates a measurable electrical signal, referred to as a peak, that is proportional to the amount of analyte present. Detector response is plotted as a function of the time required for the analyte to elute from the column after injection. The resulting plot, signal output versus time, is called a chromatogram. The results of a complete chromatographic run are printed by an integrator and strip chart recorder, or by a GC software program. The printout showing the peak (or peaks if more than one compound was present) is also referred to as a chromatogram. Detector response is generally a Gaussian shaped curve representative of the concentration distribution of the analyte band as it elutes from the column. The position of the peaks on the time axis usually serves to identify the components, and peak height, or the area under the peaks provides a quantitative measure of the amount of each component.
System Components of a Basic GC System
The primary components of a GC include:
- injection port and mobile phase
- column
- Detector(s)
- Data acquisition system
Injection Port and Mobile Phase
The mobile phase, or carrier gas as it is also known, is introduced in the injection port where the sample is volatilized and its components transformed into the gaseous phase. The carrier gas and sample move through the column, where compound separation occurs. The carrier gas and compounds eluting from the stationary phase of the chromatographic column then enter the detector. The signal from the detector then is amplified and displayed by the data system.
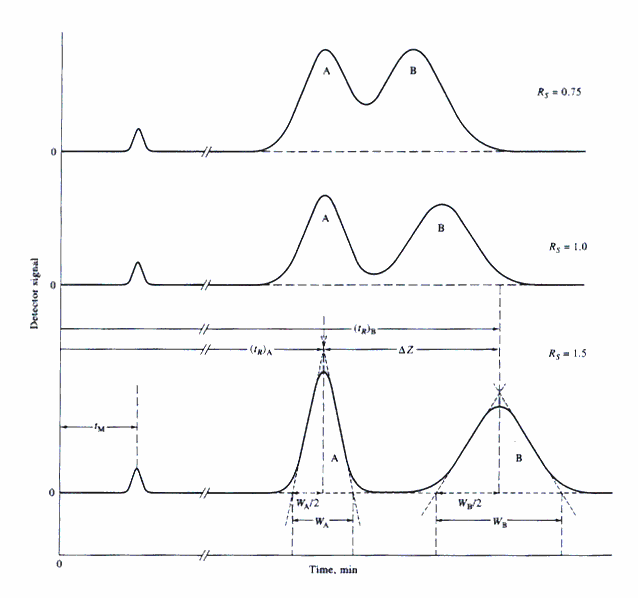
The carrier gas is generally an inert gas; often nitrogen, helium, or hydrogen. The choice of carrier gas is frequently determined by the type of detector used, and the grade of gas by how tolerant the system (column and detector) is of impurities. By increasing the speed (flow rate) of the carrier gas, the analysis time can be reduced; however, optimal peak resolution may be compromised. Peak resolution is the clean separation of chromatographic peaks. Unresolved peaks may overlap, partially or completely, compromising their identification and quantitation. A faster flow rate also sweeps the injector more efficiently, improving introduction of the sample into the column.
For those analyses that require hydrogen, this gas can be generated onsite using a portable hydrogen generator. Portable hydrogen generators can be run from an automobile cigarette lighter (12v power supply), or a transformer can be used to step-down the main voltage power supply. Hydrogen generators can supply gas for one month, giving enough carrier gas for one GC with a flame ionization detector (FID) [system example]. Having an onsite gas generator may be advantageous at sites where compressed gas cylinder storage and transport are problematic.
Column
A capillary column is an open tube made of flexible fused silica with an outer coating of durable polyimide plastic and an inner coating of stationary-phase material. The stationary phase is an organic liquid compound that is either coated on or covalently bonded to the silica interior surface of the column. The most widely used capillary columns are the fused silica, due to its strength and flexibility. The polarities of the compounds whose identities and quantities are to be evaluated dictate the choice of stationary phase, under the rule "like dissolves like." Commonly used stationary phases include:
- polydimethyl siloxane (commonly referred to as OV-1, SE-30) for PCB or PAH, separation
- polyethylene glycol (Carbowax 20M) used for free acids, alcohols, and glycols
- 50% polydimethyl siloxane (OV-17) for pesticides and glycols
- 50% trifluoropropyl-polydimethyl siloxane (OV-210) for chlorinated aromatics, nitroaromatics, and alkyl substituted benzenes
- 5% phenyl polydimethyl siloxane (OV-3 or SE-52) for halogenated organics.
Capillary columns vary in length from 15 to 100 meters, in a coiled configuration to fit in the instrument oven. For environmental analysis, 30- to 60-meter columns typically are used. Shortening the length of the column can shorten the analysis time; however, compound resolution (separation) may be compromised. If resolution is not compromised for a particular analytical application, analysis time can be reduced with a shorter column.
The diameters of open tubular capillary columns are typically between 0.75 and 0.25 millimeter, with high resolution columns having diameters of 0.20 to 0.15 millimeter. The smaller diameter columns require special injection splitting to reduce the sample size and prevent column overload. Columns referred to as mega bore open tube columns are also available and have a greater capacity, but at the expense of resolution. However, these columns have better resolution than the older type of packed columns. The smaller diameter produces better resolution and greater selectivity, but can handle only a small volume of sample (1 to 2 microliters).
A lesser used, and older column type is the packed column. Packed columns use a stainless steel or glass tube with a 2 - 4 mm inner diameter, 1 - 4 m in length, packed with a solid stationary phase. Although an old technology, packed columns have some features that may sometimes be advantageous in that they can accept high sample loading and can give a greater analytical dynamic range. Accurate quantitation and precision can be achieved by the use of a packed column, but analytical run time may be significantly prolonged. The effectiveness of a chromatographic column in separating solutes (analytes) is dependent on a number of variables. Understanding these variables is essential to the process of optimizing any chromatographic system and achieving resolution of analytes. Variables that affect separation include distribution equilibrium constants, retention time, retention (capacity) factors, and selectivity factors. A discussion of each of these is available.
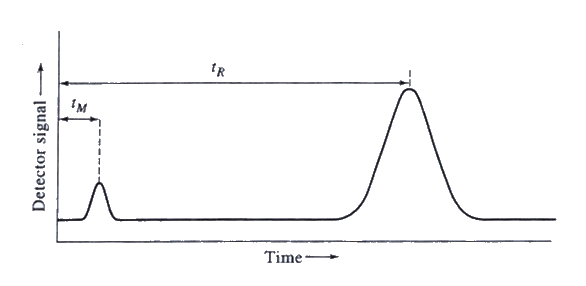


The previous discussions all assume Gaussian distributions of analytes as they elute from the column. However, non-Gaussian shaped peaks do occur and the peak shapes provide information relative to chromatographic variables. Two frequent phenomena related to non-Gaussian peak shapes that occur are fronting and tailing. With fronting, the front side of a peak is drawn out while the tail (or backside) on the right is steep. The most frequent cause of fronting is too large of a sample introduced into the column. Peak fronting due to column overload is usually accompanied by a slight shortening of retention time. The more frequent phenomenon of peak tailing results in the right side of a peak (the tail) is drawn out. This may occur when active sites in the chromatographic system strongly retain a compound. Fronting and tailing can occur when the solute has a concentration dependent (non-linear) distribution coefficient. A worn column and dead space in the GC system can also cause poor peak shape. Poor peak shape may prevent peak identification by chromatographic software, and may lead to inaccurate quantitation.
Field analysis is frequently performed on highly contaminated soils and waters, and samples can have concentrations or constituents that can ruin column performance. When this occurs, columns are replaced, often at significant expense. At this point, analysts need to insure that data produced with the new column is consistent with and comparable to data produced with the previous column. This is accomplished by analyzing quality control samples to demonstrate comparable separation and sensitivity is achieved.
Another problem associated with coated capillary columns (and not exclusive to field analysis) is column bleed. This phenomenon is best described as the elution of the stationary phase. A frequent cause of column bleed is excessive oven temperature. The end results of column bleed are extraneous peaks in the chromatogram and ultimately a fouled detector. Sensitive detectors such as the electron capture detector (ECD) are highly susceptible to column bleed. Some manufacturers of GC instruments and specialty items such as columns provide useful troubleshooting guides for common chromatographic problems. One very useful example is provided by MSP KOFEL.
Gas Chromatography Detectors
A variety of detectors for gas chromatographs are available. In general, each detector takes advantage of a unique characteristic of a molecule and uses that characteristic to generate a measurable electrical signal. A discussion of the most frequently used detectors is presented below. Principles of operation, classes of compounds providing optimal response, and detection limits are included. Click on the detector name for a schematic of the detector.
Because of the unique and complex nature of mass spectrometry, the mass spectrometer as a GC detector is discussed under the Mass Spectroscopy section.
A most useful summary of the use, capabilities and possible combinations of GC detectors is provided by the manufacturer SRI, a company that specializes in transportable GCs for field use.
Photoionization Detector
A photoionization detector (PID) consists of a special ultraviolet lamp, ranging in energy from 9.5 to 11.7 eV, mounted on a low-volume flow-through cell. As constituents of the sample pass through the cell, they are energized and ionized. The ions are collected at positively charged electrodes, where the change in current is measured.
The 10.2 eV lamp emits ultraviolet light at 121 nanometers (nm), which is sufficient to ionize BTEX compounds and hexane. A few halogenated compounds that have ionization potentials of less than 11.7 eV can be detected by the higher-energy PID. The PID is more selective than the FID.
The PID can detect VOCs (aromatic and chlorinated) and petroleum constituents including benzene, toluene, ethylbenzene and xylene (BTEX.) The PID can detect BTEX in the low ppb to high part per trillion range.
The PID is a nondestructive detector that can be used in series before other detectors. Using multiple detectors extends the range of compounds that can be detected in one analysis. PID is sensitive to humidity and may require frequent recalibration.
Flame Ionization Detector
A flame ionization detector (FID) consists of a stainless steel jet constructed so that carrier gas exiting the column flows through the jet, mixes with hydrogen, and burns at the tip of the jet. Hydrocarbons and other molecules which ionize in the flame are attracted to a metal collector electrode located just to the side of the flame. The resulting electron current is amplified by a special electrometer amplifier which converts very small currents to millivolts.
The FID is sensitive to almost all molecules that contain hydrocarbons. Examples include aromatic and chlorinated VOCs, petroleum constituents, SVOCs, and PCBs.
The FID is a destructive detector that can be used in series only after nondestructive detectors. The FID is sensitive to water, but has a wider linear range of detection than the PID. The FID also can detect more compounds than the PID.
The FID can detect compounds that in the low ppb to high part per trillion range.
Electron Capture Detector
An electron capture detector (ECD) consists of a sealed stainless steel cylinder that contains radioactive nickel-63. The nickel-63 emits beta particles (electrons) which collide with the carrier gas molecules ionizing them in the process. A stable cloud of free electrons thus forms in the ECD cell. When an electronegative molecule such as a halogenated molecule enters the cell, it immediately combines with one of the free electrons which temporarily reduces the number of free electrons. The detector electronics pulse at a variable rate to measure the electrons remaining in the cell.
The ECD is highly sensitive to electronegative molecules (those capable of producing negatively charged ions) such as halogenated compounds or those that contain nitrogen. The ECD readily detects chlorinated pesticides, halogenated solvents, and PCBs. The ECD is a nondestructive detector that can be used in series before other detectors. The ECD is sensitive to water that affects the condition of the Ni-63 foil that covers the detector. The foil must be reconditioned when its sensitivity diminishes. Because the ECD contains a radioactive source, users may be subject to licensing requirements. The Nuclear Regulatory Commission (NRC) is the agency in the United States that regulates radioactive materials. However, in certain states, the regulations are enforced by a state agency. There are two types of ECDs that require licenses; specific license ECDs and general license ECDs. A general license ECD is an ECD that customers in the United States can purchase without having their own radioactive material license. General License ECDs are typically covered under the distributor's distribution license. General License customers are required to complete and sign the General License Registration Card and comply with regulations. The owner of a General License ECD becomes a General Licensee when the ECD is purchased. The owner does not have to apply for a General License from the NRC or State Agency but could be required to register the ECD within the state. Specific License ECDs require the end user to have a Materials License from the NRC or local State Agency, which permits the owner to posses the applicable type and quantities of radioactive material. Holders of Specific Licenses have greater flexibility with their ECD and more responsibilities. For example, Specific License customers are allowed to clean ECDs and work with open sources. Distributors of ECDs are required to notify customers of licensing requirements.
The ECD can detect halogenated compounds in the low ppb to part per trillion range. The more halogenated the molecule, the more sensitive the detector is to that compound. For example, the ECD is orders of magnitude more sensitive to carbon tetrachloride than to vinyl chloride.
A fairly recent development, the pulsed discharge ECD does not require the use of Ni-63, obviating the requirements for licensing. This detector has similar sensitivity to Ni-63 based ECD.
Electrolytic Conductivity Detector
An electrolytic conductivity detector (ELCD), also known as a Hall detector, is a halogen-specific detector that operates on electrolytical conductivity principles. Organic compounds eluting from a GC column form combustion products as they are mixed with hydrogen gas over a nickel catalyst at 1,000 C in a quartz tube furnace. For example, organic chlorides form hydrogen chloride (HCl). The gas stream containing the HCl is diverted into a flowing stream of liquid, often propanol. HCl readily ionizes in the liquid, the electrolyte solvent, and changes in its electrolytic conductivity are measured by the ELCD.
The ELCD is a halogen-specific detector and readily detects chlorinated pesticides, halogenated solvents, and PCBs.
The ELCD can detect halogenated compounds in the low ppb to part per trillion range. The degree of halogenation has less effect on sensitivity than is the case with the ECD. Detection limits for compounds are similar, even though the degree of halogenation may vary. It also has a wider linear range than the ECD. The ELCD is a destructive detector that can be used in series only after nondestructive detectors. The ELCD requires a higher degree of maintenance than other detectors and cannot be recommended for field use.
However, a recent development is the "dry" ELCD (DELCD). This detector does not require nickel reaction tubes and electrolytic solvent, and it measures the halogenated species directly in the gas phase. It is more highly selective than ECD to halogenated compounds. The DELCD is rugged and amenable to field use.
Thermal Conductivity Detector
A thermal conductivity detector (TCD) consists of tiny coiled wires arranged in a Wheatstone bridge configuration. Electric current flows through the filaments making them glow hot, while carrier gas exiting the column flows past the other two filaments. The gas flow carries away excess heat, and the filaments equilibrate. When a sample compound exits the column, the thermal conductivity of the gas flowing around the filaments is changed. Therefore, the filaments get hotter and the balance of the Wheatstone bridge is altered, generating a signal that is amplified and transmitted to the data collection system.
The TCD is used to detect gaseous compounds, such as nitrogen, oxygen, and other non-hydrocarbon compounds.
The TCD is a destructive detector that can be used in series only after nondestructive detectors. The TCD has limited target analyte list. Because the TCD detects nitrogen, nitrogen cannot be used as a carrier gas.
The TCD can detect gaseous compounds in the ppm range. TCD is not used for trace analysis. Larger sample volumes are required to achieve increased sensitivity. To increase sample volume sufficiently, a large-diameter packed column must be used.
Nitrogen-Phosphorous Detector and Thermionic Ionization Detector
The nitrogen-phosphorus detector (NPD) is similar to the FID, except that the hydrogen gas flow rate is reduced to 1 to 3 milliliters per minute (ml/min), and an electrically heated thermoionic bead is positioned just above the jet orifice to receive the column effluent. Analyte molecules exiting the column collide with the hot bead, and any nitrogen or phosphorus containing molecules react with the bead's surface and liberate ions. The discharged ions are attracted to a collector electrode and an electronic amplifier is used to transmit the signal to the data processing system. The Thermionic Ionization Detector (TID) is similar in design to the NPD. The column effluent contacts the hot bead surface and any molecules containing nitro functional groups, such as trinitrotoluene, undergo a catalytic surface chemistry reaction. The resulting ions are attracted to a collector electrode and the signal they generate is amplified, and sent to a data system.
The NPD is sensitive to nitrogen and compounds that contain phosphorus. The NPD typically is used in analysis for organophosphorus pesticides. In addition, it may be used in analysis of nitroaromatics (explosives), but the TID is more suited for that purpose.
The NPD is a destructive detector that can be used in series only after nondestructive detectors. The NPD is sensitive to water that affects the condition of the thermoionic bead. The active element on the bead eventually will become depleted and require replacement.
The NPD can detect nitrogen and compounds that contain phosphorus in the ppb range. Reduced sensitivity often indicates the depletion of the active element on the thermoionic bead.
Surface Acoustic Wave Sensors (SAWS)
Surface acoustic wave sensors (SAWSs) are chemical sensors that use a piezoelectric crystal as a transducer to detect and quantitate individual chemical compounds. A transducer is defined as a device that transmits energy from one system to another. This transmission of energy frequently occurs by converting the energy to a different form. A SAWS can be defined as an acoustic device that measures changes in frequency as a result of a change in mass bound to its surface.
The SAWS operates on the principle of the piezoelectric effect. The piezoelectric effect is defined as the oscillation of a crystal, at a constant frequency characteristic of the mass and shape of the quartz crystal, when an appropriate electrical potential is applied across the face. In general, a SAWS operates by applying an alternating electric current through a piezoelectric crystal transducer, causing the crystal to vibrate. As the sensor is exposed to a sample contained in the GC column effluent, chemical compounds from the sample are deposited on the crystal surface, changing the crystal's mass and oscillation frequency. Ultimately, the change in frequency is correlated to analyte mass. When the analyte desorbs, the crystal returns to its original frequency. A SAWS with a chemically selective coating is designed to respond to a specific compound or compounds, providing qualitative identification. Other SAWS are non-selective and may respond to a number of compounds; a GC is used to separate and qualitatively identify individual compounds. The observed change in acoustic energy from the crystal provides the quantitative measurement of the concentration of the analyte.
Although the SAWS is much less sensitive to humidity than to organic compounds, water vapor also can cause a sensor to respond. Water uptake always will occur because no coating is perfectly hydrophobic. The use of adsorbents to trap water in a sample before it is introduced to the sensor can reduce the effects of humidity. Besides chemical interferences, temperature can have a direct effect on the frequency of oscillation by causing thermal expansion of the crystal and affecting the acoustic properties of the chemically selective coating. A technique that often is applied to compensate for temperature is the use of a second sensor, almost identical to the first, which is maintained at the same temperature but not exposed to the target chemical.
Target analytes include VOCs, SVOCs. PCBs, and nitroaromatic explosives.
Minimum detection limits are approximately 1 to 10 parts per million (ppm) for typical VOCs and can be reduced to 10 to 100 parts per billion (ppb) through the use of a preconcentrator.
Data Acquisition System
A data acquisition system for a GC usually has two basic components. The first component amplifies and converts the analog signal from the instrument's detector into digital data. The second component that receives the digitized signal is generally a computer, loaded with a suitable software program. Some field portable GC systems can be linked to a laptop computer and the chromatography program run from a compact disc. Chromatography software can set up the run conditions for the GC, acquire data, integrate peaks, display peaks on a computer screen, print chromatograms and run reports.
Mode of Operation
VOC Analysis
Methods of introduction into the GC system for VOCs include direct injection for ambient air and soil gas, and static headspace extraction or purge and trap extraction for soil and water.
Analytes of interest include: (1) halogenated VOCs, such as vinyl chloride, methylene chloride, trichloroethene (TCE), tetrachloroethene (PCE), trichloroethane (TCA), chloroform, carbon tetrachloride, and ethylene dibromide; (2) nonhalogenated VOCs, including the ketone solvents methyl iso-butyl ketone (MIBK), methyl ethyl ketone (MEK), and acetone; (3) aromatic compounds, including and chlorobenzenes; and (4) fuels, including BTEX and other gasoline range organics.
VOC Analysis of Ambient Air:
On site air analysis can be used to test for VOCs in stationary source testing (emission inventory), hazardous waste site testing to determine appropriate levels of personal protective equipment (PPE), fence line monitoring during remediation activities, and emergency response testing.
The preferred mode of sample collection for quick analysis is to directly draw a sample of ambient air into an onsite portable GC using an internal pump in the analytical instrumentation. Air samples also may be collected in Tedlar® bags, Summa® canisters, and on Tenax™ tubes, or using SPME devices. The use of Tedlar® bags or Summa® canisters will require that an air sample (1 to 5 ml) be withdrawn from the sample by gas-tight syringe and injected into the GC system. Air samples collected on Tenax and SPME will need desorption.
SOPs for GC methods for vapor phase organics using field portable instruments that may used for general information and guidance include:
However, the specific instruments described in these SOPs may no longer be manufactured and vendors should be consulted for information on updated systems.
The detection limits for air analysis will range considerably depending on the method of sample collection and GC system. Typical detection limits will range from 5 to 200 parts per billion by volume (ppbv). Detection limits will be considerably lower if the analytes are concentrated on some type of adsorbent material and desorbed versus directly drawn into the analytical system via a sampling pump. Analytical times for VOCs should be less than 10 minutes per sample.
An important advantage of on-site air monitoring is the quick data acquisition, which allows flexibility in decision making for on-site personnel and the project manager.
When conducting emissions testing, samples that have high moisture or acid content must be pretreated prior to analysis. Exact analyte identification may not be possible with GC for the very complex mixtures from stack emissions, due to compound coelution. It is however, possible to equip the GC with two columns, which should allow identification of components of complex mixtures.
VOC Analysis of Soil Gas:
Soil gas analysis commonly is used to identify "hot spots" or source areas of VOCs in the subsurface, and can be used to approximate the extent of a subsurface plume.
Typical sample containers for soil gas include glass bulbs, Tedlar bags, Summa® canisters, SPME devices, gas-tight syringes, 22 or 40-ml headspace vials, or passive sorbent diffusion collection devices. Because analyte concentrations are generally more dilute in soil gas samples, analysis usually requires larger sample volumes than liquid samples. Therefore, a 1 to 5 milliliters aliquot of air from the soil gas sample is injected into the GC column. In contrast, liquid sample injection volumes are usually a fraction of that volume (microliters) and contain no air. Because of the relatively large amount of air injected from soil gas samples, deterioration of the column stationary phase and oxidation of the ECD foil by oxygen in the sample is possible.
The detection limit for most VOCs is 10 nanograms per liter (ng/L). Calibrations consist of direct injection using an air standard mix or methanol-based standards. The concentration is reported in ng/L. One liter of air weighs approximately 1 gram. Therefore on a weight basis, ng/L is approximately equivalent to ppb.
The advantages of soil gas analysis are that it is rapid, inexpensive, provides real-time results, and causes minimal disturbance to the site.
One limitation of soil gas analysis is that it does not always reflect a true soil concentration. The technique is limited to high volatility and low solubility compounds. Coelution problems can occur in complex mixtures, and sample carryover or cross contamination may be a problem in highly concentrated samples. Decontamination of syringes is critical, especially for chlorinated VOCs.
VOC Analyses of Soil and Water Using Static Headspace [figure]
Water samples can be collected in 40 ml volatile organic analysis (VOA) vials or directly in a 22-ml headspace vial. Soil samples can be collected in a 4-ounce glass jar or directly into a 22-ml headspace vial. Mass or volume is measured into a headspace vial (generally 5 ml or 5 grams). No sample preparation is required for water samples. Sample preparation for soil samples may vary depending on the initial analyte concentration and soil type. For high concentration soil samples (ppm levels), studies have shown that methanol-flood (based on SW-846 method 5035) achieves the greatest extraction efficiency. An aliquot of the methanol can then be analyzed using the static headspace technique. Methanol-flood techniques, however, are not appropriate for soil samples that contain less than 200 mg/kg of an analyte due to dilution. SW-846 Method 5021 (VOC in Soils and Other Solid Matrices Using Equilibrium Headspace) recommends the addition of an aqueous matrix-modifying solution to low concentration samples. However, the addition of a preservative matrix-modifier is probably unnecessary in the case of rapid turn-round field analysis.
Static headspace extraction is widely used in determining VOCs in waste water, soil, and drinking water. This extraction method is highly productive and cost effective, requiring minimal sample preparation. Efficiency of headspace extraction is based on soil or water partition coefficients of the volatile organic analytes. The principle follows Henry's Law, where the vapor pressure of the solute in the headspace is proportional to its mole fraction in solution. In other words, when a sample containing VOCs is sealed in a headspace vial, the vapor pressure of the VOC in the headspace is proportional to its concentration in solution. This phenomenon allows for analysis of the headspace gas to determine VOC concentrations of the sample matrix without time consuming solvent extractions [Mathematical expression of Henry's Law].
The static headspace technique relies on two actions: (1) diffusion of analyte into the headspace, and (2) diffusion back into the matrix. A steady-state equilibrium is reached when the rate of analyte diffusion into the headspace is perfectly balanced by analyte diffusion back into the matrix. Low viscosity liquids reach equilibrium faster than solids. A constant heat time is recommended for samples that do not reach equilibrium within a reasonable time.
Analysis time ranges from 10 to 30 minutes depending on number and boiling point of analytes of interest.
Detection limits for BTEX compounds and most chlorinated VOCs are in the range of 1 to 10 ppb; detection limits for gasoline range purgeable compounds are 1 to 10 ppm in water and 10 to 50 ppm in soil.
Method 5021 requires the use of an automated headspace sampler. Unless a well equipped trailer is available for use, it may not be possible to employ an autosampler in the field. However, headspace analysis has been adapted for field conditions by using a gas-tight syringe to manually withdraw an aliquot of gas from the headspace above a sample and inject it into the GC.

A most informative, in-depth guide to static headspace analysis is provided by Restek.
Purge and Trap
The primary advantage of purge and trap over static headspace is that it is a dynamic process. It is a more efficient extraction technique for those VOCs which have a higher octanol/water partition coefficient, especially in soils with high organic matter content.
Purge and trap is the recommended VOC extraction technique used for GC analysis. The recommended methods are 5030A and 5035. These methods may be employed as written in a well-appointed trailer, with utilities and climate control, but both methods have been modified for use with portable field GC systems. As long as quality control on the modified method shows that the data is of known and documented quality that meets the data user's needs, modifications to SW-846 methods are acceptable.
Comparison of attributes of headspace versus purge-and-trap analysis for fixed laboratory or well equipped, trailer-based facilities.
Comparison of Attributes of Headspace Versus Purge-and-Trap Analysis
SVOC Analysis
The key in the field is to simplify the solvent extraction methods to minimize solvent waste, save time, and reduce cost. Typical solvents used in the field include hexane, methanol, and methylene chloride. Simplified field, solvent extraction methods normally do not include a cleanup step. A concentration step may also be eliminated if elevated detection limits are acceptable.
As previously noted, the Spittler soil extraction for PCBs and other semivolatile compounds is suitable for field use, and notes on this method may be found in US EPA Field Screening Methods Catalog User's Guide EPA/540/2-88/005 September 1988.
Solid-phase extraction (SPE) for water samples has some field application if facilities are available that allow for sample filtration through the extraction device, and solvent concentration. SPME is an ideal technique for field use because it is rapid, and uses no solvent. More information on SPME is available from the Sigma Aldrich company.
Thermal desorption for SVOCs is analogous to a more rigorous static headspace extraction for VOCs. It also is convenient for field use because it is simple, rapid, and requires no solvent.
Target Analytes
- Phenols
- Phthalates
- Amines
- Chlorinated pesticides
- PCBs
- PAHs
- Chlorinated hydrocarbons
- Organophosphorus compounds
- MTBE (analyze as a VOC)
- Diesel range organics
Performance Specifications
Performance specifications include information on detection limits, sample throughput, precision, accuracy, comparability to other methods, ease of deployment, ability of the instrument to complete a required number of samples.
Detection limits for GC analysis are highly dependent on the type of detector used. Parts per million (ppm) and part per billion (ppb) levels are routinely achieved with most detectors. Detection limits for each respective detector are discussed in the systems components section. Information about sample throughput, precision, accuracy, comparability to other methods, ease of deployment, ability of the instrument to complete a required number of samples may be found in EPA's Environmental Technology Verification (ETV) Reports. Examples of ETV reports are presented at the end of this document.
Quality Control
Ensuring that the data generated are of a known quality is vital to the usefulness of those data. Quality control (QC) measures take several forms. They can be performed in the field, during sample analysis, and after sample data have been collected. The type and extent of QC necessary will vary according to the test to be performed and the data quality objectives of the project. A much higher level of QC is necessary to produce defensible data that will be used alone to support specific decisions than to produce screening data that will not be used alone to support decision-making. A fuller discussion of QC for field analytical systems is presented in "Using Dynamic Field Activities for On-Site Decision Making: A Guide for Project Managers" EPA/540/R-03/002 May 2003. In addition, a comprehensive list of QC samples and the information they provide is available at:
- Summary of Quality Control Samples and the Information They Provide (77KB/4pp/PDF)
- Uniform Federal Policy for Quality Assurance Project Plans: Part 2B, Quality Assurance/Quality Control Compendium: Minimum QA/QC Activities (725KB/76pp/PDF)
Some typical field QC measures for GC systems are defined below.
Calibration - Qualitative:
Calibration consists of injecting a known compound(s) at a predetermined concentration and injection volume, and measuring the time elapsed between injection and elution. This is known as the retention time. If all variables such as temperature, flow rate, and column length are constant, the retention time of a given compound should remain consistent. To eliminate variables in injection technique, retention time relative to an internal standard can be used. Thus, if a peak appears in the chromatogram of a sample at the sample retention time of a standard, the compound is tentatively identified, not definitively. A chromatogram provides only a single piece of qualitative information about each species in a sample, its retention time. It is important to note that while chromatograms may not lead to positive identification of species present in a sample, they can provide sure evidence of the absence of a given compound (or is present at a concentration below the detection limit of the method). However, a compound can be positively identified by dual column analysis. Dual column analysis, as the name implies, requires the use of two columns with different stationary phases to identify compounds. The retention time of a particular compound will be different on each column. If a compound is positively identified on two dissimilar columns its presence in a sample is confirmed. If a compound is identified on one column, but not the other, its presence is not confirmed. However, if the organic contaminants at a site are well characterized the need for dual column confirmation is lessened.
Calibration - Quantitative:
Quantitative gas chromatography is based on comparison of either peak height or area of the analyte peak with that of one of the standards. Most modern chromatographs have digital electronic integrators that calculate accurate peak areas.
The most common method for quantitative chromatographic analysis involves the preparation of a series of standard solutions that approximate the composition of the unknown. External standard calibration generally consists of a three-point or five-point calibration. Chromatograms for the standards are generated and peak areas are plotted as a function of concentration. Criteria for acceptable calibration are described in SW 846 Method 8000B Determinative Chromatographic Separations. For the highest accuracy, frequent calibration checks are necessary to ensure that the instrument performs within acceptance criteria. At a minimum, a medium level standard (continuing calibration) is analyzed once a day, before analysis, to check the response of the instrument compared to the average response of the initial calibration. Once again, guidance for continuing calibration is given in Method 8000B. Instrument manufacturers should also be consulted to determine if there are particular requirements for any of their GC systems.
Method blanks are analyzed to check for laboratory-induced contamination and instrument blanks are analyzed to check for contamination induced by the instrument (usually by sample carryover). Method blanks are especially critical in soil gas analysis when chlorinated VOCs are target analytes.
Performance evaluation (PE) samples are spiked matrix samples that have certified concentrations of analytes and that can be purchased from reputable vendors. They are usually analyzed "blind" by the analyst (meaning the analyst does not know what analytes are present or their concentrations). The analyst must report the proper analytes and concentrations within the certified concentration ranges for the laboratory's accuracy to be acceptable. PE samples are normally not analyzed for soil gas or ambient air analysis.
Matrix spike/matrix spike duplicate (MS/MSD) samples are analyzed to check for extraction efficiency of the analytical system. Percent recoveries are calculated and must fall within an acceptable range for the extraction efficiency to be acceptable. Percent recoveries are also compared to each other by calculating relative percent differences (RPD) to assess the precision of a method. MS/MSDs are not typically run for soil gas or ambient air analysis.
Laboratory duplicates are analyzed to assess the precision of the method and homogeneity of the sample. The laboratory duplicates consist of the analysis of two aliquots of the same sample. The results from the laboratory duplicates are compared to each other through RPD calculations.
Surrogate spikes are necessary to evaluate the extraction efficiency on a per sample basis. A surrogate compound is one that is chemically similar to the target compounds but does not coelute with any of them. A percent recovery of the known spiked concentration is calculated for each sample and compared to site specific control limits. (Not typically analyzed in soil gas or ambient air samples.)
Laboratory control samples (LCS) consist of a "well-behaved" matrix spiked with a known concentration of standard purchased from a separate vendor other than the one from which the calibration standards were purchased. The percent recovery for all analytes must be within an acceptable range for the accuracy of the calibration to be acceptable.
Advantages
- A broad spectrum of organic chemical compounds can be surveyed in one analysis.
- Field portable or transportable gas chromatographic analysis can be of equal quality as fixed laboratory data when proper quality control is performed.
- Dual column analysis can also provide definitive compound identification.
- Rapid analysis provides data that can be used to enable field decision-making, expediting cleanup or characterization.
Limitations
- Coelution of analytes in single column systems.
- Instrument operation requires a higher degree of expertise than most other field instrumentation.
- Sample carryover and equipment contamination.
- Multicomponent (and therefore multipeak) compound interferences with single peak compounds, e.g.
- Petroleum with BTEX or PAH
- Aroclor 1260 with single peak chlorinated pesticides such as DDT
- Toxaphene with single peak chlorinated pesticides
Choosing a field GC and Cost Data
Points to consider in the choice if a field GC:
- Field portable, or transportable? A portable GC should be self-contained, need no exterior power source, weigh less than 20 lbs, be easily portable, and be capable of operating for a day's field work (8 hours) without consumable supplies being replenished. Transportable GCs are systems that can be packed into a crate for shipping and transported by truck or van. These instruments are not hand portable, and generally require operation from the back of a van.
- Durability. Is the GC capable of handling adverse conditions? Can it operate in conditions of high humidity, high and low ambient temperatures? Will the operator be able to use the GC when wearing mandated personal protective equipment such as gloves? Is the instrument rugged?
- Ease of operation. Are the instrument's controls easy to use when the operator's hands are cold? Are the panels that display results or operating parameters, easy to read, even in poor lighting conditions?
- Applicability. Can the instrument manufacturer customize the GC to your requirements? Does the manufacturer provide "hands-on" training on the system, or do they provide instructional videos or DVDs?
- Detection limits. Can the instrument achieve the detection limits required?
- Dynamic range. Does the GC have a wide dynamic range, reducing the need for sample dilutions?
- Turn-around. What kind of sample throughput can be expected? Is the introduction of the sample into the GC straightforward?
- Sample delivery options. Does the instrument support a variety of sampling techniques, such as loop and syringe injection, sampling by probe, SPME?
Manufacturers of Field GC Systems
Cost Data
Manufacturers of gas chromatographs occasionally establish elevated rental prices to encourage purchase rather than lease. However, scientific instrumentation suppliers provide rental gas chromatographs capable of most analyses.
Purchase price varies widely depending on instrument capabilities, auto-sampling accessories, and detectors. Simple chromatographs capable of analyzing simple mixtures of BTEX can be purchased for less than $10,000.
Lease prices also vary according to capability. Monthly rental for a simple instrument designed to analyze simple BTEX samples is about $1,500. More sophisticated instruments lease for approximately $3,000 per month.
Manufacturers listed through the link above should be contacted directly for cost information.
Verification/Evaluation Reports
Verification of the performance of site characterization and field analytical technologies is conducted through a variety of programs. Evaluation and verification reports from EPA's Superfund Innovative Technologies Evaluation (SITE) Measuring and Monitoring Program, and EPA's Environmental Technology Verification Program (ETV) program.
Superfund Innovative Technologies Evaluation (SITE) Measuring and Monitoring Program
The SITE Demonstration Program encourages the development and implementation of innovative treatment technologies for (1) remediation of hazardous waste sites and (2) monitoring and measurement. In the SITE Demonstration Program, the technology is field-tested on hazardous waste materials. Engineering and cost data on the innovative technologies are gathered so that potential users can assess the technology's applicability to a particular site. Data collected during the field demonstration are used to assess the performance of the technology, the potential need for pre- and post-treatment processing of the waste, applicable types of wastes and waste matrices, potential operating problems, and approximate capital and operating costs. The following reports from the measuring and monitoring program are available for gas chromatography:
No reports available for this technology
EPA's
Environmental Technology Verification (ETV) Program
ETV Program verifies the performance of innovative technologies. ETV was created to substantially accelerate the entrance of new environmental technologies into the domestic and international marketplaces. ETV verifies commercialized, private sector technologies. After the technology has been tested, the companies will receive a verification report that they can use in marketing their products. The results of the testing also are available on the Internet. The ETVreorts and statements listed below have been included to provide general descriptions of a particular technology rather than a information about a currently manufactured GC system.The GC systems described in these reports have been superseded by newer models, but the recent models often retain features and capabilities of the older instruments, thus the ETV reports are of continuing value. Please note that the Sentex Sentograph GC systems are now part of the Inficon company. The following reports from the ETV program are available for gas chromatography:
- Electronic Sensor Technology Model 4100 Model was verified for measurement of chlorinated volatile organic compounds in water. The verification documents available consist of a verification report (506KB/78pp/PDF) and verification statement (506KB/3pp/PDF).
- Photovac (Perkin-Elmer Corporation - Photovac Monitoring Instruments) Voyager was verified for measurement of chlorinated volatile organic compounds in water. The verification documents available consist of a verification report (599KB/84pp/PDF) and verification statement (27KB/3pp/PDF).
- Sentex Systems Scentograph Plus II was verified for measurement of chlorinated volatile organic compounds in water. The verification documents available consist of a verification
report (550KB/81pp/PDF) and verification statement (28KB/3pp/PDF).
- Joint Verification Statement (73KB/4pp/PDF) Gas Chromatography Measurement of Explosives in Contaminated Soil Model 8610C GC/TID SRI Instruments
