X-Ray Fluorescence
- Direct-Push Technologies
- Explosives
- Fiber Optic Chemical Sensors
- Gas Chromatography
- Geophysical Methods
- High-Resolution Site Characterization (HRSC)
- Immunoassay
- Infrared Spectroscopy
- Laser-Induced Fluorescence
- Mass Flux
- Mass Spectrometry
- Open Path Technologies
- Passive (no purge) Samplers for Groundwater
- Test Kits
- X-Ray Fluorescence
Description
XRF instruments are field-portable or handheld devices for simultaneously measuring metals and other elements in various media.
Typical Uses

Courtesy Olympus Corporation
Energy dispersive X-ray fluorescence (EDXRF) is a method of detecting metals and other elements, such as arsenic, selenium, bromine, and iodine, in soil and sediment. Some of the primary elements of environmental concern that EDXRF can identify are arsenic, barium, cadmium, chromium, copper, lead, mercury, selenium, silver, and zinc. Most instruments can be made to detect any element between atomic weight 19 (potassium) and 92 (uranium). Some will detect 11 (sodium) through 94 (plutonium), with lighter elements (atomic weight 11 to 18) 11 to 18 having higher detection limits than heavier elements. The handheld or field-portable (FPXRF) units use techniques that have been developed for analysis of numerous environmental contaminants in soil and sediment. They provide data in the field that can be used to identify and characterize contaminated sites and guide remedial work, among other applications. An eight-part August 2008 XRF Web seminar is available with supporting materials in the CLU-IN Seminar Archive. The course manual ![]() is available for a January 2010 8-hour classroom course on XRF applications, XRF-specific QA/QC and data use.
is available for a January 2010 8-hour classroom course on XRF applications, XRF-specific QA/QC and data use.
FPXRF analyzers also have found use in identifying surfaces coated with paints that were cut with polychlorinated biphenyls (PCBs). A high chlorine content, which is generally not found in paints, can be indicative of PCBs. The use of PCBs in paints occurred in the 1950s through 1970s until they were banned. Older military activities, and Department of Energy facilities, utilities, and other industrial sites may have this problem.
Air particulate matter can be collected on a filter using a low- or high-volume sampler. The filter can then be removed and the elemental content measured by XRF. Appendix Q ![]() in 40 CFR 50 contains a (June 1999) method of how this is done for lead PM10 filter analysis.
in 40 CFR 50 contains a (June 1999) method of how this is done for lead PM10 filter analysis.
FPXRF analyzers also can be used to detect metals in water. Trace metals are rarely found in water at concentrations FPXRFs can detect. Because water quenches FPXRF emissions, the return X-ray signal will likely be weak; therefore, the ions to be measured need to be taken out of the aqueous phase and concentrated to achieve detection limits in the low parts per billion (ppb) range that are applicable for maximum contaminant levels (MCLs). Several methods can be used:
- The water sample is filtered and concentrated on an ion exchange membrane, which is dried and analyzed by FPXRF (Potts and West 2008).
- Agar is used to collect the ions, and the water sample is filtered. The agar will form a thin membrane that can be dried and analyzed by FPXRF (Nakano et al. 2009).
- Ammonium pyrrolidinedithiocarbamate can be added to the water sample to precipitate target metals. The sample is then filtered and the dried precipitate analyzed by FPXRF (IAEA 2007).
- The water sample is concentrated using a rotary evaporator and a pipet is used to drop the concentrated fluid onto an Ultra Carry® support film (Moriyama et al. 2004).
Theory of Operation
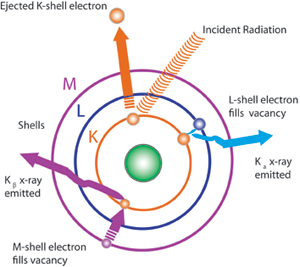 In FPXRF analysis, a process known as the photoelectric effect is used in analyzing samples. Fluorescent X-rays are produced by exposing a sample to an X-ray source that has an excitation energy similar to, but greater than, the binding energy of the inner-shell electrons of the elements in the sample. Some of the source X-rays will be scattered, but a portion will be absorbed by the elements in the sample. Because of their higher energy level, they will cause ejection of the inner-shell electrons. The electron vacancies that result will be filled by electrons cascading in from outer electron shells; however, since electrons in outer shells have higher energy states than the inner-shell electrons they are replacing, the outer shell electrons must give off energy as they cascade down. The energy is given off in the form of X-rays, and the phenomenon is referred to as X-ray fluorescence. Because every element has different electron shell energies, each element emits a unique X-ray at a set energy level or wavelength that is characteristic of that element. The elements present in a sample can be identified by observing the energy level of the characteristic emission X-rays. The intensity of the emission X-rays is proportional to the concentration and can be used to perform quantitative analysis.
In FPXRF analysis, a process known as the photoelectric effect is used in analyzing samples. Fluorescent X-rays are produced by exposing a sample to an X-ray source that has an excitation energy similar to, but greater than, the binding energy of the inner-shell electrons of the elements in the sample. Some of the source X-rays will be scattered, but a portion will be absorbed by the elements in the sample. Because of their higher energy level, they will cause ejection of the inner-shell electrons. The electron vacancies that result will be filled by electrons cascading in from outer electron shells; however, since electrons in outer shells have higher energy states than the inner-shell electrons they are replacing, the outer shell electrons must give off energy as they cascade down. The energy is given off in the form of X-rays, and the phenomenon is referred to as X-ray fluorescence. Because every element has different electron shell energies, each element emits a unique X-ray at a set energy level or wavelength that is characteristic of that element. The elements present in a sample can be identified by observing the energy level of the characteristic emission X-rays. The intensity of the emission X-rays is proportional to the concentration and can be used to perform quantitative analysis.
The K, L, and M shells are those commonly involved in FPXRF analysis. A typical emission pattern, or emission spectrum, for a given element has several peaks generated from the emission of X-rays from those shells. Each of these shells also has alpha and beta emissions at different energies. For example, K alpha spectra for lead are around 74.96 kev, while K beta are found at 84.92 kev. L alpha for lead are around 10.55 and L beta 12.61kev. Note that since XRF affects inner shell and not bonding electrons, the XRF spectrum of an element is independent of its chemical form. See the Table for individual element Kev values for K and L shell electrons.
System Components
An XRF analyzer consists of three major components: (1) a source that generates X-rays (a radioisotope or X-ray tube); (2) a detector that converts X-rays emitted from the sample into measurable electronic signals; and (3) a data processing unit that records the emission or fluorescence energy signals and calculates the elemental concentrations in the sample. The following sections describe each of the components in greater detail.
- Radioisotopes
An X-ray source will excite characteristic X-rays from an element only if the source energy is greater than the binding energy, or absorption edge energy, of the electrons in a given electron shell. A given individual radioisotope source can only excite certain elements. Analysis is more sensitive for an element with an absorption edge energy similar to, but less than, the excitation energy of the source.
Because individual sources by nature reliably analyze only a limited number of elements, FPXRF instruments that use more than one source have been developed, allowing them to address a greater number and range of elements. Typical arrangements of such multisource instruments include Cd-109 and Am- 241 or Fe-55, Cd-109, and Am-241.
XRF units that employ radioisotope sources are regulated by the states and require a license to operate. In addition, these units have special shipping requirements. One reason for the regulation and shipping requirements is the radioisotope sources are always generating x-rays whether they are in use or not.
- X-ray Tube Sources
Miniature X-ray tube sources are the excitation technique of choice for new equipment and are replacing radioisotope sources in most environmental applications. Because it only generates x-rays when it is being used, an advantage of the X-ray tube source is that it does not generally require licensing or special shipping, as do XRF units employing radioisotope sources; however, some states require registration of the equipment.
In an X-ray tube, electrons are accelerated in an electrical field and shot against a target material where they are decelerated. The technical means of achieving this is to apply high voltage (typically up to 50 kV for portable equipment) between a heated cathode (e.g., a filament) and a suitable anode material (Schlotz and Uhlig 2006). The anode is usually made with silver or rhodium but can be constructed using other elements depending upon the specific use of the instrument. Note that some elements (e.g., lead, mercury, uranium) have K shell electrons whose excitation energy is greater than 50 keV; for these elements, the X-ray tube sources will only produce L lines. The X-ray spectrum generated by an X-ray tube includes the characteristic lines of the anode materials plus Bremsstrahlung radiation. Filters can be used to tailor the source profile for better detection limits.
Two basic types of detectors are used in FPXRF units: gas-filled and solid-state. Each detector has its advantages and limitations and is better suited to some applications than to others.
Gas-filled proportional counters are generally not used in field-portable environmental XRF instruments.
Resolution, given in electron volts (eV), is a measure of a detector's ability to separate energy peaks. Some elements produce peaks that are near each other in the spectrum. Very high concentrations of one element may produce a peak that overwhelms the nearby peaks of other elements that are present at lower concentrations. The higher the resolution (i.e., the lower the eV number), the better the detector is able to separate characteristic peaks. The XRF operator must be careful to select an FPXRF unit that has sufficient resolution to satisfy the data quality needs of the project.
Common solid-state detectors include Si(Li), HgI2, silicon pin diode, and silicon drift detector (SDD). Among these detectors, the SDD, which was introduced most recently in handheld instruments, is capable of the highest resolution (120-139 eV) and has the greatest counting rate. The Si(Li) has a resolution of about 170 eV if cooled to at least �90°C, either with liquid nitrogen or by thermoelectric cooling that uses the Peltier effect. The HgI2 detector can operate at a moderately subambient temperature and is cooled by use of the Peltier effect. It has a resolution of 270 to 300 eV. The silicon pin diode detector operates near ambient temperatures and is cooled only slightly by use of the Peltier effect. It has a resolution of 145 eV to 180 eV.
Each manufacturer has a software package to convert spectral data into concentration results as determined from factory calibration data, sample thickness as estimated from source backscatter, and other parameters (Palmer 2011). The instrument may also have a fundamental parameters mode, which is used when the elements of concern are expected to be at percent concentration levels (e.g., metal alloy identification and scrap yard applications). The software package is usually set up to also allow the user to generate site-specific calibration data. The software permits downloading data, including spectral data, to a PC for further evaluation. Manufacturers often set their software packages to look for a specific array of elements. Always ensure that all the project's contaminants of concern are included in the software package set.
Mode of Operation
To perform an analysis, a sample is positioned in front of the plastic film measurement window of the probe, and measurement of the sample is initiated, usually by depressing a trigger or start button. Doing so exposes the sample to the X-ray radiation. The length of time the sample is exposed is referred to as the count time. Fluorescent and backscattered X-rays from the sample reenter the analyzer through the window and are counted by the instrument's detector. X-rays emitted by the sample at each energy level are called "counts." The detector records the counts, measures the energy of each X-ray, and builds a spectrum of element peaks on a multichannel analyzer (MCA). The unit's software integrates the peaks to produce a measurement of concentrations of elements and, usually, the standard deviation for each analyte based on the counting statistics. At the completion of the exposure time, the instrument software statistically computes a concentration from the readings collected from each energy level along the spectrum.
Count times from 30 seconds per source to as long as 600 seconds per source can be employed, depending on the data quality needs of the project. As count times increase, the detector collects a larger number of X-rays from the sample, including more X-rays from elements that are present at comparatively lower concentrations. In general, a 4x increase in count time results in a 2x decrease in detection limits, although there will be diminishing returns beyond several hundred seconds. Also, the longer the count time, the greater the reading's precision (but not necessarily accuracy).
In Situ and Intrusive Analysis
An FPXRF detector can be operated in the in situ or the intrusive mode. The particular requirements of the job, such as the required detection limits or data sample precision and accuracy, and the purpose of sampling—for field screening or for quantitative analysis—will determine which mode is appropriate and what count times are needed.
- In Situ
XRF instrument in situ analysis refers to the rapid screening of soil in place and is generally used to locate hotspots. For in situ operation, the window of the probe is placed in direct contact with the surface to be analyzed and a trigger is pulled. Because analyses in this mode typically are completed very quickly (in less than one minute) and heterogeneity of the samples (both in terms of matrix and element concentration) can be a concern, it is recommended to take three to four measurements in a small area and then average the values to determine the concentrations of target elements. Care should be taken to ensure good contact with the sample (i.e., not on uneven surfaces, rocks, sticks, etc.) to limit x-ray backscatter and measurement variability in this mode.
- Intrusive
Intrusive analysis is used to ensure greater precision and accuracy when lower detection limits are needed. These goals are achieved through more extensive sample preparation and longer analysis times to reduce heterogeneity among samples and increase the sensitivity of the instrument, respectively. For intrusive operation, a sample is collected, prepared (usually by homogenizing, drying, grinding, and sieving), and placed in a sample cup that has a transparent Mylar® or polyester film window. The sample cup is placed under the probe window (some units provide a safety cover for intrusive analysis) and analyzed. Handheld FPXRF instruments can analyze samples in either mode, while transportable instruments are generally limited to intrusive samples. Preparation of samples is discussed in greater detail in the performance specifications section.
Target Analytes
Instruments with X-ray tubes and silicon drift detectors are capable of analyzing sodium through plutonium elements (generally excluding the noble gases) with varying detection limits. Note that low voltage (40-50 kV) X-ray tubes may limit the query to L lines in high Z elements (Barium and higher, Palmer undated 2011) Also, some instruments have the capability to create a vacuum or use a helium purge system to eliminate air in the space in front of the detector, which will lower its detection limits. This ability is very useful for lower-weight elements (e.g., magnesium and aluminum). Note that the software and emission source of a given instrument may be targeted toward a much smaller range of elements.
The radioisotope sources that have been standard in FPXRF units are Fe-55, Cd-109, Am-241, and Cm-244. Elements that those sources are commonly used to analyze include:
| Source | Activity (mCi) |
Half-Life (Years) |
Excitation Energy (keV) |
Elemental Analysis Range |
|---|---|---|---|---|
| Fe-55 | 20-50 | 2.7 | 5.9 | Sulfur to Chromium K Lines Molybdenum to Barium L Lines |
| Cd-109 | 5-30 | 1.3 | 22.1 and 87.9 | Calcium to Rhodium K Lines Tantalum to Lead K Lines Barium to Uranium L Lines |
| Am-241 | 5-30 | 432 | 26.4 and 59.6 | Copper to Thulium K Lines Tungsten to Uranium L Lines |
| Cm-244 | 60-100 | 17.8 | 14.2 | Titanium to Selenium K Lines Lanthanum to Lead L Lines |
Performance Specifications
Performance specifications include information about interferences, detection limits, calibration, sample preparation, quality control, and precision and accuracy.
Interferences
A variety of factors can affect the detection and quantification of elements in a sample. Some factors can be prevented or minimized through careful preparation and sample design; others are natural effects that must be taken into consideration. To produce useful data, it is important that the analyst understand the relevant interferences.
- Elemental Matrix Effects
The presence of different elements and their concentrations in a sample can lead to X-ray absorption and enhancement phenomena. For example, iron tends to absorb copper X-rays and will interfere with low levels of chromium, and high levels of lead will interfere with low levels of arsenic. The effects can be corrected mathematically through the FPXRF instrument's software. See the Table for individual element Kev values for K and L shell electrons.
- Physical Matrix Effects
Matrix effects can cause a great deal of variation in sample analyses. Because XRF is predominantly a surface measurement (X-rays penetrate a few millimeters at most into soil), the physical matrix of the sample is important. Physical matrix effects result from variations in the physical character of the sample soils, such as particle size, uniformity, homogeneity, and condition of the surface. If the data quality objective is to provide an accurate quantitative concentration, every effort should be made to homogenize the soil samples thoroughly before analysis. A discussion of this process is given in the sample preparation section.
- Moisture Effects
Moisture content above 20 percent may cause problems because moisture alters the soil matrix for which the FPXRF has been calibrated. Moisture can also have a dampening effect on the return signal. These problems can be minimized by drying., preferably in a convection or toaster oven. Drying by microwave is not recommended because it can increase variability between the FPXRF data and confirmatory data and can cause arcing if fragments of metal are present in the sample (EPA SW-846). Also, if a comparison between FPXRF data and confirmatory data is made, ensure to account for results reported on a wet or dry weight basis, especially when comparing to ICP.
Sampling Effects
In environmental samples, typical X-ray penetration depths range from 0.1 millimeter (mm) to 1 mm. Inconsistent positioning of samples in front of the probe window is a potential source of error because the X-ray signal decreases as the distance from the source increases. Maintaining a consistent distance between the window and the sample minimizes that problem. For best results, the window of the probe should be in direct contact with the sample or material housing the sample, such as mylar or poly films over cups, or plastic bag.
Detector Resolution Effects
The resolution of the detector may cause problems in analyzing some elements. If the energy difference between the characteristic X-rays of two elements (as measured in eV) is less than the resolution of the detector in eVs, the detector will not be able to resolve the peaks. In other words, if two peaks are 240 eVs apart, but the resolution of the detector is 270 eV, the detector will have difficulty in differentiating those peaks. A common example is the overlap of the arsenic Ka peak with the lead La peak. With the use of mathematical corrections that subtract an estimated La lead interference, lead can be measured from the lead Lb peak, and arsenic still can be measured from the arsenic Ka peak. The concentration of arsenic will be less precise, however, because of the statistical estimation of the lead La concentration.
Detection Limits
An FPXRF operator must consider two types of detection limits: instrument detection limits (DL) and method detection limits (MDL). A DL is the absolute threshold concentration of a given element that a particular instrument can resolve, as determined by the standard deviation (SD) of an individual analytical result. Instruments equipped with SDDs can achieve DLs of <5 to <50 ppm for most elements between potassium and plutonium (e.g., <5 ppm for zinc, lead, and mercury and <10 ppm for chromium, copper, and cadmium).
MDLs depend on the analytical method (such as preparation and analysis times) and sample matrix and may be higher than DLs. The results of replicate measurements of a low-concentration sample can be used to generate an average site-specific MDL. The MDL is defined as three times the SD of the results for a replicate analysis of a low-concentration sample. XRF users should be aware that many instrument configurations will report a measured value (as a function of counting statistics) that is less than 3 SDs of the counting statistics as a non-detect. In some instances it may be of value to use a "detected" number even if it is less than 3 SDs (e.g., statistical evaluations of the data). In these instances it may be worth while to talk with the manufacturer about whether these numbers can be made available.
Calibration
FPXRF units are calibrated by any of several methods. The methods will vary according to the make of the unit and the intended use of the data, such as for screening or for quantitative analysis.
- Fundamental Parameters Calibration
The fundamental parameters (FP) calibration is a "standardless" calibration. Rather than calibrating a unit's calibration curve by measuring its response to standards that contain analytes of known concentrations, FP calibration relies on the known physics of the spectrometer's response to pure elements to set the calibration. Built-in mathematical algorithms are used to adjust the calibration for analysis of soil samples and to compensate for the effects of the soil matrix. The FP calibration is performed by the manufacturer, but the analyst can adjust the calibration curves (slope and y-intercept) on the bases of results of analyses of check samples, such as standard reference materials (SRM), which are analyzed in the field. This mode is best for samples containing elements that can be detected by XRF (i.e., alloys, well characterized samples, and samples containing relatively high concentrations of elements) (Palmer 2011).
- Empirical Calibration
In performing an empirical calibration, a number of actual samples, such as site-specific calibration standards (SSCS), are used, and the instrument's measurements of the concentrations of known analytes in the samples are taken. Empirical calibration is effective because the samples used closely match the sample matrix. SSCSs are well-prepared samples collected from the site of interest in which the concentrations of analytes have been determined by a reference method, such as inductively coupled plasma, atomic absorption, or other methods approved by the regulatory agency. The standards should contain all the analytes of interest as well as interfering analytes. Manufacturers recommend the use of 10 to 20 calibration samples to generate a calibration curve. When comparing the methods for calibration, take note of the acid digestion-spike recoveries (efficiency) of the analytical method to assure that the lab method data is compatible with the XRF data. Poor efficiency means that a direct comparison of the data will be difficult. Total digestion (e.g. SW 846 Method 3052) can be expensive and may not be the best analytical value if a 1:1 calibration is insisted upon. One solution is using a reference method along with the XRF method in a collaborative fashion. For a discussion of laboratory versus field determined values see Section 2.7 of ITRC 2003.

- Compton Normalization
The Compton normalization method incorporates elements of both empirical and FP calibration. A single well-characterized standard, such as an SRM or SSCS, is analyzed, and the data are normalized for the Compton peak. The Compton peak is produced from incoherent backscattering of X-ray radiation from the excitation source and is present in the spectrum of every sample. The intensity of the Compton peak changes as various matrices affect the way in which source radiation is scattered. For that reason, normalizing to the Compton peak can reduce problems with matrix effects that vary among samples. Compton normalization is similar to the use of internal standards in analysis for organic analytes.
- Sample Preparation
Procedures for sample preparation for in situ and intrusive analysis vary considerably because the two methods serve completely different purposes. Sample preparation for in situ analysis is fairly straightforward, while sample preparation for intrusive analysis can be fairly complicated, depending on the data quality required.
In situ or "point-and-shoot" analysis requires little sample preparation and is generally used for field screening of hotspots:
- First, any unrepresentative debris (e.g., rocks, pebbles, leaves, vegetation, roots) should be removed from the surface of the soil.
- Second, the surface must be smooth to allow the probe window to make good contact with the soil surface.
- Finally, the surface of the soil should not be saturated to the point that ponded water is present.
For an "in situ prepared" sample, which provides for better quantitation and detection limits:
- Soil from the sampling point is collected, and all unrepresentative debris (e.g., rocks, pebbles, leaves, vegetation, roots) is removed.
- The soil is placed in a plastic bag and thoroughly homogenized. At a minimum, this should include mixing and breaking of aggregate material to best obtain a uniform particle size.
- The sample probe is placed directly on the bag for analysis. In very rare cases, plastic bags can have potential interference (often attributable to bag construction [ribbed, overly thick, or crinkled tend to increase x ray scatter and variability] and moisture within the bag). This potential for interference should be checked by taking repeated measurements of NIST standards and/or well characterized site samples with and without shooting through the bags.
- When performing the analysis, a series of measurements is recommended to understand sample heterogeneity and to generate an average concentration. For example, one could use two or more measurements on the front side of the bag followed by two or more on the back side of the bag.
For intrusive analysis, the sample first must be collected and then prepared for analysis in a sample cup or bag. Some or all of the following steps may be necessary, depending on the data quality needed:
- Any large, unrepresentative debris should be removed from the sample.
- The most important preparation step is thorough homogenization. Mixing the sample in a plastic bag and breaking any aggregate material to best obtain a uniform particle size works well. If a high degree of precision and accuracy is not required, the sample can be analyzed in the bag. A representative bag should be tested to determine if it contains any trace elements of concern and if it interferes with emission or return X-rays. Testing can be done by taking repeated measurements of NIST standards and/or well characterized site samples with and without shooting through the bags.
- If the sample contains more than 20 percent moisture, the sample should be dried, preferably in a convection or resistive oven. Drying in a microwave oven is discouraged because doing so can increase the variability between the FPXRF data and confirmatory data, and arcing can occur when metal fragments are present in the sample. Note that mercury is volatile, and if it is a suspected contaminant of concern, the samples should be air dried.
- If a high degree of precision and accuracy is required or if a particular soil fraction is of interest (e.g., fines <60 mesh to evaluate dust inhalation), the sample should be passed through a sieve. If the sample is not wet (has a moisture content of less than 20 percent) and is not high in clay content, the sample can be sieved in the field before it is placed in a container; otherwise, the sample is ground with a mortar and pestle or ball mill and passed through a 40- or 60-mesh sieve after drying.
- Finally, the sample is placed in a 31- or 40-mm polyethylene cup and covered with Mylar film.
- Quality Control
Ensuring that the data generated by FPXRF analysis are of a known quality is vital to ensuring the usefulness of those data, regardless of their purpose. Quality control (QC) measures take several forms and can be performed in the field, during sample analysis, and after sample data have been collected. The amount and type of QC necessary will depend on the project's data quality objectives. A higher degree of QC is necessary to produce defensible, definitive data, but analytical results from intrusive analysis have been demonstrated to compare favorably with results obtained through traditional laboratory methods, given that sample preparation has been thorough and QC adequate. By nature, results obtained in situ are of lower quality because of the lack of sample preparation. A typical QC program might include the following measures:
- An energy calibration check sample.
- An instrument blank.
- A method blank.
- A calibration verification check sample, which is usually accomplished through a series of NIST standards like 2709b, 2710b, and 2711b.
- A precision sample.
- A confirmatory sample (Note: Be sure to specify that the analysis is for total metals, not extractable.).
Advantages
Most field-transportable instruments weigh less than 30 pounds and can be operated using battery power for 8 to 10 hours. Handheld instruments generally weigh less than 5 pounds.
Depending upon the amount of sample preparation required, a relatively large number (25 to 150) of analyses can be done in a day.
Analyses of as many as 25-plus elements can be performed simultaneously in a single analysis.
The sample is not destroyed during preparation or analysis; therefore, it is possible to perform replicate analyses on a sample and send the same sample for confirmatory analysis so that comparability studies can be performed. The sample also can be archived for later use as a soil standard.
Because no solvents or acids are used for sample extraction, little waste is generated; disposal costs therefore are reduced.
Operators usually can be trained in one or two days. The software is menu-driven. No data manipulation is required. Instruments are marketed for use by general scientists.
With proper preparation, results can be obtained that are comparable to offsite laboratory values.
Limitations
While detection limits for elements of concern using an X-ray tube and SDD are often below applicable screening action levels, matrix effects may cause the actual method detection limit to be higher. Full intrusive preparation techniques can result in significantly lower method detection limits, but also increase the cost of analysis.
Concentrations of elements in different types of soil or matrices might change, causing potential interferences—for example, between arsenic and lead. Site-specific calibration standards can compensate for some of those effects.
A specific license is required to operate FPXRF instruments with isotopic sources. The total cost of attending a radiation safety course, obtaining the necessary paperwork, and paying the fee for the license can range from $500 to $1,000.
Any instrument that has a Si(Li) detector will require liquid nitrogen and a dewar (aluminum container) to hold the liquid nitrogen. Obtaining and handling liquid nitrogen to cool an instrument with a Si(Li) detector before analysis can be performed adds to project time and cost.
Cost Data
XRF costs vary significantly ($25,000 to $50,000). Instrument design and accessories affect instrument prices. Manufacturers should be contacted directly for cost information. Also, instruments can be rented, and some consulting firms offer FPXRF surveys as a service.
Resources
Advanced Design Application and Data Analysis for FP-XRF in Soil Matrices, 2010 North American Environmental Field Conference and Exposition![]()
CERCLA Education Center. 2010.
Incremental-Composite Sampling (ICS) and XRF: Tools for Improved Soil Data![]()
Crumbling, D., U.S. EPA Office of Superfund Remediation and Technology Innovation, Technology and Field Services Division. 2013.
Inorganic Compendium Method IO-3.3: Determination of Metals in Ambient Particulate Matter
Using X-Ray Fluorescence (XRF) Spectroscopy![]()
EPA 625-R-96-010a, 1999.
Introduction to Energy-Dispersive X-Ray Fluorescence (XRF): An Analytical Chemistry Perspective![]()
Palmer, P., San Francisco State University.
Analytical Sciences Digital Library, 79 slides, 2011.
Method 6200: Field Portable X-Ray Fluorescence Spectrometry for the Determination of
Elemental Concentrations in Soil and Sediment![]()
U.S. EPA, Test Methods for Evaluating Solid Waste, Physical/Chemical Methods, SW-846
An Overview of XRF Basics![]()
PSchlotz, R. and S. Uhlig.
Bruker AXS GmbH, 2006.
Portable X-Ray Fluorescence Spectrometry: Capabilities for In Situ Analysis
Potts, P.J. and M. West.
RSC Publishing, 291 pp, 2008.
PPB Analysis by XRF: A New Micro-Droplet Method for Environmental Liquid Sample (Abstract)![]()
Moriyama, T., Y. Yamada, H. Inoue, J.E. Martin, and H. Kohno.
2004 Denver X-Ray Conference, August 2-6, 2004, Steamboat Springs, CO.
Preconcentration of Environmental Waters by Agar for XRF Analysis (Abstract)![]()
Nakano, K., K. Okubo, and K. Tsuji.
Powder Diffraction 24(2):135(2009).
XRF Newsletter![]()
International Atomic Energy Agency (IAEA), No. 14, Dec 2007.
Additional Information
Contamination, Validation & Verification of Mercury Impacted Soil: ADI Mulwala![]()
Australian Government, Department of Defence, 110 pp, 2004.
Draft Bruker XRF Spectroscopy User Guide: Spectral Interpretation and Sources of Interference![]()
Kaiser, B. and A. Wright.
Bruker AXS GmbH, 53 pp, 2008.
Expedited Characterization and Excavation of Lead in Soil Using X-Ray Fluorescence
(XRF), Ross Metals Site, Rossville, TN.
U.S. EPA, Triad Resource Center, 2004.
Field-Portable X-Ray Fluorescence (FP-XRF) Determinations of Metals in Post-Blast Ordnance Residues![]()
Walsh, M.E.
ERDC/CRREL TR-04-5, 33 pp, 2004.
First Research Co-ordination Meeting under Co-ordinated Research Project on In Situ Applications
of XRF Techniques![]()
International Atomic Energy Agency (IAEA), IAEA/AL/130, 91 pp, 2001.
Operator of Portable X-ray Fluorescence Analyzers: Certification Information and
Examination Preparation Booklet
Natural Resources Canada, Government of Canada, Version 3, 2010.
Sample Handling Strategies for Accurate Lead-in-Soil Measurements in the Field and Laboratory![]()
Shefsky, S.
International Symposium of Field Screening Methods for Hazardous Wastes and Toxic Chemicals,
Las Vegas, Nevada, January 29-31, 1997.
XRF CLU-IN Sessions
Verification/Evaluation Reports
- Superfund Innovative Technologies Evaluation (SITE) Measuring and Monitoring Program
The former SITE Demonstration Program encouraged the development and implementation of innovative treatment technologies for (1) remediation of hazardous waste sites and (2) monitoring and measurement. In the SITE Demonstration Program, the technology was field-tested on hazardous waste materials. Engineering and cost data on the innovative technologies were gathered so that potential users could assess the technology's applicability to a particular site. Data collected during the field demonstration were used to assess the performance of the technology, the potential need for pre- and post-treatment processing of the waste, applicable types of wastes and waste matrices, potential operating problems, and approximate capital and operating costs. Funding for this program was discontinued. SITE demonstrations for XRF performed in 2006 can be found on line. The equipment tested in these demonstrations may still be in use, but the particular models are no longer produced. All of the demonstrated models used radioisotopes as sources.
- EPA's Environmental Technology Verification (ETV) Program
EPA's Environmental Technology Verification (ETV) Program verifies the performance of innovative technologies. ETV was created to substantially accelerate the entrance of new environmental technologies into the domestic and international marketplaces. ETV verifies commercialized private-sector technologies. After the technology has been tested, the companies receive a verification report that they can use in marketing their products. The results of the testing also are available on the Internet. Two XRF instruments were evaluated in 1996 and four were evaluated in 1998. These evaluations are dated and do not represent the capabilities of newer equipment. The evaluations can be found on line.
